Summary
Clinical characteristics.
Fructose-1,6-bisphosphatase (FBP1) deficiency is characterized by episodic acute crises of lactic acidosis and ketotic hypoglycemia, manifesting as hyperventilation, apneic spells, seizures, and/or coma. Acute crises are most common in early childhood; nearly half of affected children have hypoglycemia in the neonatal period (especially the first 4 days) resulting from deficient glycogen stores. Factors known to trigger episodes include fever, fasting, decreased oral intake, vomiting, infections, and ingestion of large amounts of fructose.
In untreated individuals, symptoms worsen progressively as continued catabolism leads to multiorgan failure (especially liver, brain, and later heart). Morbidity and mortality are high. Sepsis, blindness, and Reye syndrome-like presentation have been reported.
In between acute episodes, children are asymptomatic. While the majority of affected children have normal growth and psychomotor development, a few have intellectual disability, presumably due to early and prolonged hypoglycemia.
Diagnosis/testing.
The diagnosis of FBP1 deficiency is established in a proband with suggestive clinical and metabolic findings by identification of EITHER biallelic FBP1 pathogenic variants on molecular genetic testing OR deficient fructose-1,6-bisphosphatase 1 (FBP1) activity in liver or mononuclear white blood cells. Molecular genetic testing is generally preferred because of its widespread availability and accuracy.
Management.
Treatment of manifestations: Intervention (oral or IV glucose) should take place early in an acute crisis while the blood glucose is normal due to the possibility of delayed hypoglycemia, which only occurs relatively late in the course of acute metabolic decompensation.
The mainstay of routine daily management is prevention of hypoglycemia by avoiding fasting (including use of uncooked cornstarch overnight), consuming frequent meals, and appropriate management of acute intercurrent illnesses.
Prevention of primary manifestations: Routine daily management to prevent hypoglycemia, attention to agents/circumstances to avoid, and routine immunizations, including annual influenza vaccine to reduce the risk of infection, which can precipitate hypoglycemia.
Surveillance: Long-term monitoring of developmental milestones in affected children and quality of life issues for affected individuals and their parents/caregivers; monitoring for excessive weight gain at each visit.
Agents/circumstances to avoid: Food items or medicines that contain fructose, sucrose, glycerol, and/or sorbitol, especially during acute crisis in infancy or early childhood. Although small amounts of fructose (≤2 g/kg/day) are generally well tolerated, single ingestion of high dose of fructose (>1g/kg) is harmful, especially in younger children. Fructose tolerance testing ("fructose challenge") to diagnose FBP1 deficiency can be hazardous and should not be performed.
Evaluation of relatives at risk: When the familial FBP1 variants are known, it is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk sibs of an affected individual in order to identify as early as possible those who would benefit from prompt initiation of preventive measures.
Pregnancy management: For a pregnant woman with FBP1 deficiency, consider referral to a high-risk obstetric center and consultation with a metabolic physician. Home glucose monitoring and consumption of uncooked cornstarch at night as needed as carbohydrate requirements increase during pregnancy. During labor, continuous glucose infusion is recommended to maintain euglycemia.
Diagnosis
Formal diagnostic criteria for fructose-1,6-bisphosphatase (FBP1) deficiency have not been established.
Suggestive Findings
FBP1 deficiency should be suspected in individuals with the following characteristic clinical features and metabolic findings.
Clinical features
Episodes of acute crisis may manifest as hyperventilation, apneic spells, seizures, and/or coma, most commonly in neonates and infants. The course of illness is precipitous and may be lethal, especially in neonates and infants.
Other features can include episodic irritability, tachycardia, hypotonia, and hyperhidrosis.
Hepatomegaly is common.
Inter-crisis periods are uneventful with normal development and growth.
Metabolic findings. A strong clinical suspicion should arise in individuals with recurrent episodes of ketotic hypoglycemia with lactic acidosis and urinary organic acid analysis showing a glycerol and glycerol-3-phosphate peak. Although this combination is fairly specific for FBP1 deficiency, establishing the diagnosis requires enzyme assay and/or molecular genetic testing (see Establishing the Diagnosis).
Hypoglycemia (plasma glucose <40mg/dL in neonates; <60 mg/dL in older infants, children, and adults; reference range 70-120 mg/dL)
High anion-gap metabolic acidosis
Lactic acidemia (plasma lactate >2.5 mmol/L; reference range: 0.5-2.2 mmol/L) with possible elevated lactate:pyruvate ratio and hyperalaninemia.
Ketosis (individuals with low ketones have been reported)
Elevated glycerol 3-phosphate is an important biomarker for this disorder on urine organic acid analysis. Glycerol 3-phosphate is more specific as glycerol is also elevated in glycerol kinase deficiency (see
NROB1-Related Adrenal Hypoplasia Congenita) [
Nakai et al 1993]. It is important to collect the urine sample in the period of crisis, as the glycerol 3-phosphate level returns to normal once the individual is in the well state [
Moey et al 2018].
Pseudo-hypertriglyceridemia. Although plasma triglycerides are commonly increased, it is not the triglycerides but the glycerol levels that are high (often referred to as "pseudo-hypertriglyceridemia"). The commonly available biochemical assays for plasma triglycerides cannot differentiate glycerol from triglycerides, and hence overestimate the triglyceride levels due to the high concentration of glycerol in plasma [
Afroze et al 2013]. Glycerol blanking methods can be used to measure the true levels of triglycerides [
Cole 1990].
Hyperuricemia (plasma uric acid >5.0 mg/dL; reference range 2.0-5.0 mmol/L
Increased free fatty acids (in some cases)
Establishing the Diagnosis
The diagnosis of fructose-1,6-bisphosphatase 1 deficiency is established in a proband with suggestive clinical and metabolic findings by identification of EITHER biallelic FBP1 pathogenic (or likely pathogenic) variants on molecular genetic testing (Table 1) OR deficient fructose-1,6-bisphosphatase 1 (FBP1) activity in liver or mononuclear white blood cells. Molecular genetic testing is generally preferred because of its widespread availability and accuracy.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic FBP1 variants of uncertain significance (or of one known FBP1 pathogenic variant and one FBP1 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular Genetic Testing
When the clinical and metabolic findings suggest the diagnosis of fructose-1,6-bisphosphatase deficiency, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
Note: Pathogenic variants reported frequently in individuals of certain ancestries are noted in
Table 7. Given the rarity of fructose-1-6-bisphosphatase deficiency, the efficacy of targeted testing for specific pathogenic variants in individuals of these ancestries is not known.
A multigene panel (such as one for hypoglycemia or lactic acidosis) that includes
FBP1 and other genes of interest (see
Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of
uncertain significance and pathogenic variants in genes that do not explain the underlying
phenotype. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. Of note, given the rarity of fructose-1,6-bisphosphatase deficiency, some panels may not include
FBP1. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused
exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see
Table 1).
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Table 1.
Molecular Genetic Testing Used in Fructose-1,6-Bisphosphatase Deficiency
View in own window
| Gene 1 | Method | Proportion of Pathogenic Variants 2 Detectable by Method |
|---|
|
FBP1
| Sequence analysis 3 | ~90% 4 |
| Gene-targeted deletion/duplication analysis 5 | ~10% 6 |
- 1.
- 2.
- 3.
- 4.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
Analysis of Fructose-1,6-Bisphosphatase (FBP1 or FBPase) Activity
While enzymatic activity in leukocytes and liver is very specific, testing is not widely available [Besley et al 1994, Lebigot et al 2015]: spectrophotometric analysis to estimate enzyme activity in leukocytes is performed in a few laboratories worldwide. The usual assay tests FBP1 activity by measuring the amount of NADPH formed – a nonspecific assay as NADPH may be formed from other reactions within the cells. Specificity can be achieved by testing for FBP1 activity in the presence and absence of AMP, a specific inhibitor for the enzyme [Lebigot et al 2015]. FBP1 stability in untreated blood samples at +4 °C lasts 24 hours, and therefore samples can be transported long distances to a reference laboratory.
Clinical Characteristics
Clinical Description
The manifestations of fructose-1,6-bisphosphatase (FBP1) deficiency are generally episodic due to lactic acidosis and ketotic hypoglycemia, which are often triggered by fasting or febrile infections. The episodes of acute crisis are most frequent in early life – neonatal period, infancy, and early childhood – and subsequently decrease in frequency. In FBP1 deficiency, several metabolic derangements can occur with or without hypoglycemia.
Classically, FBP1 deficiency manifests in the first year of life. Nearly half of affected infants present within the first four days of life with an acute crisis. Neonatal presentation results from hypoglycemia due to deficient glycogen stores [Steinmann & Santer 2016].
Acute crises are characterized by episodes of hyperventilation, apneic spells, seizures, and/or coma, often with hepatomegaly. Muscular hypotonia may also be present. This may be associated with transient liver dysfunction (transaminitis), which does not require specific treatment [Bhai et al 2018]. With age, the frequency of attacks decreases and episodes are characterized by irritability, somnolence, hypotonia, and dyspnea.
Factors known to trigger episodes include fever, fasting, decreased oral intake, vomiting, infections, and ingestion of large amounts of fructose. Episodes tend to be recurrent. Often four to five episodes occur before the correct diagnosis is established [Lebigot et al 2015].
In between crises, children are asymptomatic and the majority experience normal growth and psychomotor development [Steinmann & Santer 2016]. A few children with brain injury and/or intellectual disability have been reported, probably related to early and prolonged hypoglycemia [Li et al 2017, Moey et al 2018].
In untreated individuals, symptoms worsen progressively as continued catabolism leads to multiorgan failure (especially liver, brain, and later heart). Morbidity and mortality are high. Sepsis, blindness, and Reye syndrome-like presentation have been reported [Lebigot et al 2015, Bhai et al 2018].
Nomenclature
Baker & Winegrad [1970] first described deficiency of hepatic fructose-1,6-bisphosphatase 1 (hence the eponymous Baker-Winegrad disease) in a child with hypoglycemia and metabolic acidosis on fasting.
Prevalence
Fructose-1,6-bisphosphatase deficiency is rare. Approximately 150 affected individuals have been reported to date.
An estimated prevalence of fructose-1,6-bisphosphatase deficiency is 1:350,000 in the Dutch population [Visser et al 2004] and <1:900,000 in the French population [Lebigot et al 2015]. The disorder may be more frequent in populations with higher rates of consanguinity [Santer et al 2016].
Differential Diagnosis
Table 2a provides a comparative analysis of disorders with clinical similarities to fructose-1,6-bisphosphatase (FBP1) deficiency. Table 2b compares the biochemical parameters of these disorders.
Information on mitochondrial respiratory chain and Krebs cycle disorders and fatty acid oxidation defects follows Table 2b.
Table 2a.
Disorders (and Associated Genes) of Interest in the Differential Diagnosis of Fructose-1,6-Bisphosphatase Deficiency
View in own window
| Gene(s) | Disorder 1 | Differential Diagnosis Disorder: Features Overlapping w/FBP1 Deficiency | Distinguishing Between FBP1 Deficiency & Differential Diagnosis Disorder |
|---|
|
ACAT1
| Beta-ketothiolase deficiency
(OMIM 203750) | Ketolytic defect characterized by ketotic hypoglycemia or hyperglycemia & metabolic acidosis | In BKD: ↑ of specific metabolites on urine organic acids by GCMS can include 2-methylacetocetate, 2-methyl-3-hydroxybutyryl CoA, & tiglylglycine. |
|
ALDOB
| Hereditary fructose intolerance 2 | When weaned onto sucrose- or fructose-containing foods, infants can manifest nausea, bloating, vomiting, sweating, abdominal pain, & growth restriction. Chronic liver & renal disease occur in untreated children.
| Overall, HFI has a more chronic course than FBP1D. Children w/HFI have strong aversion to sweets & often have renal tubular dysfunction, (not seen in FBP1D). Children w/FBP1D do not have GI symptoms or FTT w/chronic fructose ingestion.
|
G6PC1
SLC37A4
| Glycogen storage disease type I 3 | Accumulation of glycogen & fat in liver & kidneys, resulting in hepatomegaly & renomegaly Untreated infants present at age 3-4 mos w/hepatomegaly, lactic acidosis, hyperuricemia, hyperlipidemia, hypertriglyceridemia, &/or hypoglycemic seizures.
| Detection of glycerol in FBP1D (on urine organic acid analysis) is useful in differentiating the disorders. |
|
PC
| Pyruvate carboxylase deficiency 4 | Episodes of acute vomiting, tachypnea, & acidosis are usually precipitated by metabolic stress or infection; episodes may be very similar to FBP1D (w/↑ lactate-to-pyruvate ratio, hyperalaninemia, hypoglycemia, & metabolic acidosis). | The neurologic involvement, severe ID, & recurrent seizures characteristic of PCD types A & B are not observed in FBP1D. |
|
PGM1
| PGM1-CDG
(See Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview.) | Presents more commonly w/rhabdomyolysis; however, episodic hypoglycemia & metabolic acidosis may also occur. | Short stature, birth defects (incl cleft palate, bifid uvula), & dilated cardiomyopathy may be present in PGM1-CDG. Transferrin isoforms are abnormal & aid in diagnosis of PGM1-CDG.
|
| Multiple | Fatty acid oxidation
defects (FAODs) | FAODs can present in neonates w/hypoglycemia, hyperammonemia, & ↓/absent ketones. |
|
| Multiple | Mitochondrial respiratory chain disorders & Krebs cycle disorders | Multisystem involvement in which the most metabolically active organs are most affected (e.g., brain, liver, kidney, heart) | Mitochondrial disorders have a more chronic course than FBP1D. |
BKD = beta-ketothiolase deficiency; CDG = congenital disorder of glycosylation; FBP1D = FBP1 deficiency; FTT = failure to thrive; HFI = hereditary fructose intolerance; ID = intellectual disability; MS/MS = tandem mass spectrometry; PCD = pyruvate carboxylase deficiency
- 1.
Disorders included in this table are inherited in an autosomal recessive manner with the exception of mitochondrial respiratory chain and Krebs cycle disorders, which may be inherited in an autosomal recessive manner, an autosomal dominant manner, or by maternal inheritance.
- 2.
Hereditary fructose intolerance is due to deficiency of enzyme aldolase B, which facilitates the breakdown of fructose-1-phosphate into dihydroxyacetone phosphate and glyceraldehyde.
- 3.
GSDI is due to the deficiency of enzyme glucose-6-phosphatase.
- 4.
Pyruvate carboxylase enzyme aids in the irreversible carboxylation of pyruvate to oxaloacetate.
Table 2b.
Biochemical Parameters of Disorders in the Differential Diagnosis of Fructose-1,6-Bisphosphatase Deficiency
View in own window
| Biochemical Parameter | FBP1 | BKT | HFI | GSDI | PCD | FAOD | Respiratory
Chain Defects |
|---|
| ↑ lactate | Fasting | During crisis | Fasting | Permanent, also increased w/fasting | Permanent | Fasting | Permanent |
Lactate/
pyruvate ratio | 20-40 | – | – | – | >30 | – | >20 |
| Ketosis | ↑↑/– |
↑↑
|
↑
| ↑↑/– |
↑↑↑
| – |
↑↑
|
| Triglycerides | Pseudo-
hyper |
↑
| N |
↑
| N | N | N |
| Glucose | L | L | L | N/L | L/N/H | N/L | L |
| Ammonia | N |
↑
| N | N |
↑↑
|
↑
| N/↑↑ |
| Alanine |
↑↑
| N |
↑↑
| N |
↑↑
| N |
↑↑
|
| Citrulline | N | N | N | N | H | N | N |
| Liver dysfunction (transaminitis) | ↑/– |
↑
|
↑
|
↑
|
↑
| N | ↑/– |
| Uric acid |
↑
| N | N |
↑
| N | N | N |
| Organic acids in urine | Ketonuria, glycerol, glycerol-3-phosphate | 2-methylacetocetate, 2-methyl-3-hydroxybutyryl CoA, tiglylglycine | Ketonuria | Ketonuria | Lactate/
ketonuria | ↑ C16-C22 | Lactate |
↑ = increased; ↑↑ = moderately increased; ↑↑↑ = severely increased; BKT = beta-ketothiolase deficiency; FAOD = fatty acid oxidation defect; GSDI = glycogen storage disease type 1; H = high; HFI = hereditary fructose intolerance; L = low; N = normal; PCD = pyruvate carboxylase deficiency
Fatty acid oxidation defects (FAODs) are mitochondrial disorders caused by defective beta-oxidation of fatty acids. FAODs can present in neonates with hypoglycemia, hyperammonemia, and reduced/absent ketones. As ketones could be normal in FBP1 deficiency, diagnosis of FAOD is based on elevated acylcarnitine profiles on MS/MS.
Mitochondrial respiratory chain and Krebs cycle disorders. Mitochondrial respiratory chain disorders are a heterogeneous group of disorders that share involvement of the cellular bioenergetic machinery due to molecular defects affecting the mitochondrial oxidative phosphorylation system. The variable presentation can include neonatal metabolic acidosis with increased lactate. The usual disease course is a gradually progressive loss of developmental milestones; however, more rapid decline occurs with episodic crises. Urine organic acid profile may reveal distinctive elevation of fumaric acid or other Krebs cycle intermediates, reflecting the site of the enzyme deficiency. See Mitochondrial Disorders Overview.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in a child diagnosed with fructose-1,6-bisphosphatase (FBP1) deficiency who is not in acute crisis, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended [Wang et al 2017, Bhai et al 2018].
Note that the management of possible multisystem complications resulting from early and prolonged hypoglycemia are not discussed in this GeneReview.
Table 3.
Recommended Evaluations Following Initial Diagnosis of Fructose-1,6-Bisphosphatase Deficiency for a Child Not in Acute Crisis
View in own window
| Evaluation/Concern | Comment |
|---|
| Consultation w/metabolic physician / biochemical geneticist & specialist metabolic dietician | Consider obtaining baseline blood gas, lactate, ketones, CK. 1 Assess growth parameters (height, weight, head circumference). Assess diet & nutritional status.
|
| Hepatomegaly | Abdominal ultrasound to assess for hepatomegaly Baseline liver function tests Baseline serum lipid panel 2 Baseline serum uric acid
|
| Developmental assessment | Referral to developmental pediatrician to assess motor, adaptive, cognitive, & speech/language skills; need for early intervention / special education |
| Consultation w/clinical geneticist &/or genetic counselor | Incl genetic counseling |
| Family support/resources | Assess:
|
- 1.
Elevation of CK has been noted in at least one individual in acute crisis [Bhai et al 2018]. This may indicate rhabdomyolysis secondary to energy deficiency.
- 2.
Treatment of Manifestations
An international survey of 126 patients from 36 centers revealed widely varying practices of fructose/sucrose restriction; the authors concluded that internationally accepted guidelines for management and surveillance were needed [Pinto et al 2018].
Management of acute crises. Acute crises are more frequent in early life – neonatal period, infancy, and early childhood – with gradual decrease afterwards.
Intervention (oral glucose or IV dextrose) should take place early in an acute crisis while the blood glucose is normal due to the possibility of delayed hypoglycemia, which only occurs relatively late in the course of acute metabolic decompensation.
The family must be given the contact information for an expert metabolic center and a clear emergency outpatient treatment plan during illness (Table 4) and emergency letters/cards with information on principles of acute in-patient treatment (Table 5).
Emergency outpatient treatment. The threshold for emergency outpatient evaluation and treatment should be low. Acute management guidelines are available from the British Inherited Metabolic Disease Group. See protocol (pdf).
Laboratory evaluations during acute illnesses should include pH and blood gases, glucose (laboratory and bedside strip test), urea and electrolytes, complete blood count, lactate, blood culture, and urine ketones.
The child should receive emergency treatment if the child is vomiting, complaining of stomach pain, or not wanting to eat or drink, irrespective of associated symptoms (e.g., fever, cough, cold).
Table 4.
Emergency Outpatient Treatment in Individuals with Fructose-1,6-Bisphosphatase Deficiency
View in own window
Manifestation/
Concern | Treatment | Considerations |
|---|
Mildly ↑ catabolism
(fever, cough, or
cold; loose stools;
continues to eat;
not assoc w/lethargy)
| - 1.
Restriction of fructose, sucrose, glycerol, & sorbitol - 2.
↑ frequency of carbohydrate feedings - 3.
Intake of glucose polymers
|
|
|
Ketonuria
| | An early indicator of impending crisis |
Acute in-patient treatment. Acute manifestations of hypoglycemia (lethargy, encephalopathy, seizures, hyperhidrosis), often occurring in the setting of intercurrent illness and/or inadequate caloric intake, should be managed symptomatically and with generous caloric support in a hospital setting, with aggressive treatment and supportive care of any identified or clinically suspected acute conditions.
Table 5.
Acute In-Patient Treatment in Individuals with Fructose-1,6-Bisphosphatase Deficiency
View in own window
Manifestation/
Concern | Treatment | Considerations |
|---|
|
Hypoglycemia
| - 1.
IV glucose bolus (2 mL/kg of 10% dextrose) followed by continuous infusion of glucose at high rates (10% dextrose infusion) - 2.
Transition to oral/enteral feeds as clinically tolerated
| The symptoms of acute illness typically subside soon after administration of IV dextrose & child should recover quickly (w/in hrs), usually w/no residual damage. |
|
Metabolic acidosis
| - 1.
IV glucose bolus as above - 2.
If pH remains <7.1 or worsens, administer NaHCO3 as 1/2 the calculated dose over a 30-min period. - 3.
Restrict fructose, glycerol, sucrose, & sorbitol.
|
|
|
Hepatomegaly & ↑ transaminases
| None | Transient findings that resolve spontaneously |
IV = intravenous; NaHCO3 = sodium bicarbonate
Prevention of Primary Manifestations
The main principles of routine daily management are the following:
Prevention of hypoglycemia (by avoiding fasting and consuming frequent meals). Slowly absorbed carbohydrates such as uncooked cornstarch (1-2 g/kg) mixed in milk or water at bedtime may be introduced to children after age six to 12 months who have nocturnal hypoglycemia [
Pinto et al 2018].
Appropriate management of acute intercurrent illnesses, which can exacerbate the need for glucose
Restriction of food items or medicines that contain fructose, sucrose, glycerol, and/or sorbitol
Also advised are routine immunizations, including annual influenza vaccine, to reduce the risk of infections that can precipitate hypoglycemia.
Surveillance
No formal guidelines for long-term surveillance for individuals with FBP1 deficiency exist. Table 6 summarizes long-term surveillance based on the natural history of the disorder.
Table 6.
Recommended Long-Term Surveillance for Individuals with Fructose-1,6-Bisphosphatase Deficiency
View in own window
Manifestation/
Concern | Evaluation | Frequency/Comment |
|---|
|
Delayed acquisition of developmental milestones
| Monitor developmental milestones. | At each visit |
| Neuropsychological testing using age-appropriate standardized assessment batteries | As needed |
| Standardized quality-of-life assessment tools for affected individuals & parents/caregivers | As needed |
|
Excessive weight gain
| Measurement of growth, weight, & head circumference | At each visit |
Agents/Circumstances to Avoid
Avoid ingestion of food items or medicines that contain fructose, sucrose, glycerol, and/or sorbitol, especially during acute crises in infancy or early childhood.
Although small amounts of fructose (≤2 g/kg/day) are well tolerated by individuals with FBP1 deficiency, single ingestion of high doses of fructose (>1g/kg) is harmful, especially in younger children.
The fructose tolerance testing ("fructose challenge") to diagnose FBP1 deficiency can be hazardous and should not be performed.
Evaluation of Relatives at Risk
It is appropriate to clarify the status of apparently asymptomatic older and younger at-risk sibs of an affected individual in order to identify as early as possible those who would benefit from prompt initiation of measures to prevent acute crises (frequent feedings and avoidance of fasting) and prompt treatment of infections / febrile illnesses. Evaluations should include targeted molecular genetic testing if the FBP1 pathogenic variants in the family are known.
For newborns who may be at risk for FBP1 deficiency, it is important to avoid fasting, to feed regularly, and to monitor for hypoglycemia and acidosis while awaiting results of biochemical and/or molecular genetic testing. Monitoring for hypoglycemia can include use of bedside glucometers and clinical monitoring for lethargy, poor intake, vomiting, hypothermia, and/or tachypnea. A low threshold for starting an intravenous infusion of 5%-10% dextrose may help prevent triggering of an acute crisis, especially in newborns who have low birth weight, are preterm, or have a diabetic mother.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Although successful pregnancies have been reported in women with FBP1 deficiency [Åsberg et al 2010], certain precautions should be taken:
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Fructose-1,6-bisphosphatase (FBP1) deficiency is inherited in an autosomal recessive manner.
Parents of a proband
Sibs of a proband
Offspring of a proband. Unless an affected individual's reproductive partner also has FBP1 deficiency or is a carrier, offspring will be obligate heterozygotes (carriers) for one of the FBP1 pathogenic variants identified in the proband.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an FBP1 pathogenic variant.
Carrier Detection
Molecular genetic testing. Carrier testing for at-risk relatives requires prior identification of the FBP1 pathogenic variants in the family.
Note: Biochemical enzyme-based testing is not reliable for carrier detection.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the FBP1 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Note: Biochemical testing is not a reliable method for prenatal diagnosis as FBP1 enzyme activity has been reported to be low in human placenta [Papamarcaki & Tsolas 1991].
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Fructose-1,6-Bisphosphatase Deficiency: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
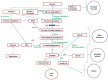
Fructose-1,6-bisphosphatase 1 (FBPase, or FBP1) is a key enzyme of the gluconeogenic pathway. Its deficiency impairs glucose production from all gluconeogenic precursors, including dietary fructose. As a result, euglycemia in persons with FBP1 deficiency depends on glycogen stores in the liver. During glycemic stress (including catabolic states such as infections and fasting), glycogen stores are depleted and the gluconeogenic substrates pyruvate, alanine, and glycerol accumulate (). The subsequent conversion of pyruvate into lactate and acetyl CoA results in lactic acidemia and ketosis.
Pathophysiology of FBP1 deficiency
Deficient conversion of glucose-1-phosphate to glucose also activates the pentose phosphate pathway, causing both the production of ribose-5-phosphate and the synthesis of purines and pyrimidines. The subsequent breakdown of purines and pyrimidines results in hyperuricemia.
Mechanism of disease causation. Loss of fructose-1,6-bisphosphatase 1 function
FBP1-specific laboratory technical considerations.
FBP1 comprises one noncoding exon (exon 1) and seven coding exons; it spans more than 31 kb [el-Maghrabi et al 1995]. Deletions of exons 2, 3, 3-7, and 8 as well as whole-gene deletions have been reported.
(The six FBP1 introns are in the same positions in the rat gene.)
Table 7.
Notable FBP1 Pathogenic Variants
View in own window
| Reference Sequences | DNA Nucleotide
Change | Predicted
Protein Change | Comment [Reference] |
|---|
|
NM_000507.3
| c.-24-26_170+5192 (deletion exon 2) | -- | Observed in the homozygous state in affected persons in Armenia & Turkey [Santer et al 2016] |
NM_000507.3
NP_000498.2
| c.472C>T | p.Arg158Trp | Frequently observed in France, India, Pakistan, Saudi Arabia, & Brazil [Lebigot et al 2015, Ijaz et al 2017, Bhai et al 2018, Moey et al 2018, Pinheiro et al 2019] |
| c.611_614delAAAA | p.Lys204ArgfsTer72 | Observed in India & Pakistan [Ijaz et al 2017, Bhai et al 2018] |
| c.685C>T | p.Glu229Ter | Common in Morocco [Prahl et al 2006, Lebigot et al 2015] |
| c.841G>A | p.Glu281Lys | Common in India, Pakistan, & Saudi Arabia [Afroze et al 2013, Santer et al 2016, Ijaz et al 2017, Bhai et al 2018] |
| c. 959dupG | p.Ser321ValfsTer13 | Common in Japan; observed in China, Korea, & in North Americans & Europeans [Kikawa et al 1997, Herzog et al 2001, Takagi et al 2013, Lee et al 2019] |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.