Epimerase Deficiency Galactosemia
Synonyms: Galactosemia Type III, GALE Deficiency, UDP-Galactose-4'-Epimerase Deficiency
Judith Fridovich-Keil, PhD, Lora Bean, PhD, Miao He, PhD, and Richard Schroer, MD.
Author Information and AffiliationsInitial Posting: January 25, 2011; Last Update: March 4, 2021.
Estimated reading time: 29 minutes
Summary
Clinical characteristics.
Epimerase deficiency galactosemia (GALE deficiency galactosemia) is generally considered a continuum comprising several forms:
Generalized. Enzyme activity is profoundly decreased in all tissues tested.
Peripheral. Enzyme activity is deficient in red blood cells (RBC) and circulating white blood cells, but normal or near normal in all other tissues.
Intermediate. Enzyme activity is deficient in red blood cells and circulating white blood cells and less than 50% of normal levels in other cells tested.
Infants with generalized epimerase deficiency galactosemia develop clinical findings on a regular milk diet (which contains lactose, a disaccharide of galactose and glucose); manifestations include hypotonia, poor feeding, vomiting, weight loss, jaundice, hepatomegaly, liver dysfunction, aminoaciduria, and cataracts. Prompt removal of galactose/lactose from their diet resolves or prevents these acute symptoms. Longer-term features that may be seen in those with generalized epimerase deficiency include short stature, developmental delay, sensorineural hearing loss, and skeletal anomalies. In contrast, neonates with the peripheral or intermediate form generally remain clinically well even on a regular milk diet and are usually only identified by biochemical testing, often in newborn screening programs.
Diagnosis/testing.
The diagnosis of epimerase deficiency galactosemia is established in a proband with impaired GALE activity in RBC or other cells and/or biallelic pathogenic variants in GALE identified on molecular genetic testing. The degree of GALE enzyme activity impairment in RBC does not distinguish between the clinically severe generalized and the milder intermediate or peripheral forms of epimerase deficiency; further enzymatic testing in other cell types such as stimulated leukocytes or EBV-transformed lymphoblasts is required to make that distinction.
Management.
Treatment of manifestations: The common acute and potentially lethal symptoms of generalized epimerase deficiency galactosemia are prevented or corrected by a galactose/lactose-restricted diet. Note: Affected individuals may require trace environmental sources of galactose: infants should be fed a formula (e.g., soy formula) that contains trace levels of galactose or lactose. Continued dietary restriction of dairy products in older children is recommended. In contrast, infants with peripheral epimerase deficiency galactosemia are believed to remain asymptomatic regardless of diet; infants with intermediate epimerase deficiency galactosemia may benefit in the long term from early dietary galactose/lactose restriction, but this remains unclear. Standard treatment for developmental delay, skeletal anomalies, poor weight gain / failure to thrive, mature cataracts, and sensorineural hearing loss.
Prevention of primary manifestations: In generalized epimerase deficiency galactosemia, restriction of dietary galactose/lactose appears to correct or prevent the common acute signs and symptoms of the disorder (hepatic dysfunction, renal dysfunction, and mild cataracts), but not the developmental delay or learning impairment observed in some affected individuals. Because of the difficulty in distinguishing peripheral and intermediate forms of epimerase deficiency galactosemia, dietary restriction of galactose/lactose is recommended for all infants with GALE deficiency, relaxing the restriction, as warranted, once a more accurate diagnosis has been confirmed.
Surveillance: Hemolysate gal-1P (galactose-1-phosphate) or urinary galactitol is monitored, especially if the diet is to be normalized. Acceptable levels of RBC gal-1P are not known, but are estimated to be <3.5 mg/100 mL (normal ≤1.0 mg/100 mL) on data from classic galactosemia. Other parameters that warrant monitoring are growth and developmental milestones, vision, and hearing (particularly in those in whom hearing loss has been identified).
Agents/circumstances to avoid: Dietary galactose/lactose in persons with generalized epimerase deficiency galactosemia, certainly as infants and perhaps for life.
Evaluation of relatives at risk: Each at-risk newborn sib should be treated with dietary restriction of galactose from birth while awaiting results of diagnostic testing for epimerase deficiency galactosemia; either molecular genetic testing (if the pathogenic variants in the family are known) or measurement of GALE enzyme activity in RBC (if the pathogenic variants in the family are not known) can be performed.
Genetic counseling.
Epimerase deficiency galactosemia is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a GALE pathogenic variant, each full sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk family members, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible if the pathogenic variants in the family are known.
Diagnosis
Epimerase deficiency galactosemia (GALE deficiency galactosemia) is a continuum comprising three forms:
Generalized. Enzyme activity is profoundly decreased in all tissues tested.
Peripheral. Enzyme activity is deficient in red blood cells (RBC) and circulating white blood cells, but normal or near normal in all other tissues.
Intermediate. Enzyme activity is deficient in RBC and circulating white blood cells and less than 50% of normal levels in other cells tested.
Suggestive Findings
Epimerase deficiency galactosemia should be suspected in individuals with the following newborn screening results, suggestive clinical features, and supportive laboratory findings while on a normal milk diet.
Newborn screening results
Suggestive clinical features
Supportive laboratory findings in an infant drinking breast milk or a dairy milk formula
Elevated RBC hemolysate gal-1P concentration (normal: 0-1.0 mg/100 mL RBC):
Urinary galactose concentrations as high as 116 mmol/L (2.09 g/100 mL, control <30 mg/100 mL)
Non-glucose reducing substance in the urine (which represents urinary galactose)
Elevated urinary galactitol concentrations (normal: <94.7 mmol/mol creatinine for age <1 year, <45.3 mmol/mol creatinine for age 1-6 years, <18.4 mmol/mol creatinine for age >6 years)
Generalized aminoaciduria
Normal GALT, GALK, and GALM enzyme activities
Note: If epimerase deficiency galactosemia is suspected, assessment of enzymatic activity for GALT, GALK, and GALM is
not required prior to pursuing either measurement of GALE enzyme activity or
molecular genetic testing for
GALE (see
Establishing the Diagnosis). If Gal-1P is elevated, GALT activity should be tested to rule out classic galactosemia, but GALT testing may be done concurrently with GALE. GALM activity testing may not be clinically available.
Establishing the Diagnosis
A diagnosis of epimerase deficiency galactosemia is established in a proband by ONE OR MORE of the following:
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic GALE variants of uncertain significance (or of one known GALE pathogenic variant and one GALE variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular testing approaches can include single-gene testing or a multigene panel:
A multigene panel that includes
GALE and other genes of interest (see
Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of
uncertain significance and pathogenic variants in genes that do not explain the underlying
phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused
exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Table 1.
Molecular Genetic Testing Used in Epimerase Deficiency Galactosemia
View in own window
| Gene 1 | Method | Proportion of Pathogenic Variants 2 Detectable by Method |
|---|
|
GALE
| Sequence analysis 3 | 14/16 alleles and 13/14 alleles (~90%) 4 |
| Gene-targeted deletion/duplication analysis 5 | None reported 6 |
- 1.
- 2.
- 3.
- 4.
Whole-gene sequencing has revealed ostensibly causal GALE variants in most persons with biochemically confirmed GALE deficiency who have been studied (e.g., Park et al [2005], Openo et al [2006], reviewed in Berry et al [2020]); however, due to the small number of alleles studied and the biochemical complexity of the diagnosis this estimate may change with time.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
At the time of this writing, no deletions or duplications involving GALE have been reported to cause epimerase deficiency galactosemia.
Additional Testing
GALE enzyme activity can be measured in fibroblasts or lymphoblasts to help distinguish between the generalized, peripheral, and intermediate forms of epimerase deficiency galactosemia; however, to the authors' knowledge this testing is not currently offered on a clinical basis.
Clinical Characteristics
Clinical Description
The clinical severity of epimerase deficiency galactosemia caused by reduced activity of the enzyme GALE [Berry et al 2020] ranges from potentially lethal [Holton et al 1981, Henderson et al 1983, Walter et al 1999, Sarkar et al 2010] to apparently benign [Gitzelmann 1972].
Epimerase deficiency galactosemia can be divided by apparent enzyme activity level in specific cell types into the following three forms: generalized, peripheral, and intermediate (see Diagnosis) [Openo et al 2006]. Note: In all three forms, GALE enzyme activity is deficient in peripheral circulating red and white blood cells.
A key difference between generalized epimerase deficiency galactosemia and intermediate or peripheral epimerase deficiency galactosemia is that individuals with generalized epimerase deficiency galactosemia develop clinical findings on a normal milk diet, while infants with peripheral or intermediate epimerase deficiency galactosemia remain clinically well, at least in the neonatal period.
Generalized Epimerase Deficiency Galactosemia
Generalized epimerase deficiency galactosemia is rare, with only nine individuals from five families described in the literature [Holton et al 1981, Garibaldi et al 1983, Henderson et al 1983, Sardharwalla et al 1988, Walter et al 1999, Sarkar et al 2010, Dias Costa et al 2017]. summarizes the key clinical features of individuals reported with this phenotype.
Table 2.
Generalized Epimerase Deficiency Galactosemia: Frequency of Select Features
View in own window
| Feature | # of Persons
w/Feature 1 | Comment |
|---|
| Hepatic abnormalities | 8/9 | May be ameliorated by treatment 2 |
| Short stature | 7/7 | In those who survive the neonatal period |
| Developmental delay | 6/6 | Incl both children who experienced acute illness before diagnosis & younger sibs who were switched to a low-galactose diet before onset of severe neonatal symptoms |
| Hypotonia | 6/8 | May be ameliorated by treatment 2 |
| Sensorineural hearing loss | 4/7 | |
| Micrognathia | 4/6 | |
| Flexion deformities of the fingers | 3/6 | |
| Hip dysplasia | 3/7 | |
| Cataracts | 3/8 | May be ameliorated by treatment 2 |
| Renal dysfunction/tubulopathy | 1/6 |
- 1.
Not all affected individuals were assessed for each feature listed.
- 2.
Infants with generalized epimerase deficiency galactosemia who are on a diet containing galactose/lactose typically present with symptoms reminiscent of classic galactosemia: hypotonia, poor feeding, vomiting, weight loss, jaundice, hepatomegaly, liver dysfunction (e.g., markedly elevated serum transaminases), aminoaciduria, and cataracts. Renal dysfunction was also noted in one affected individual [Dias Costa et al 2017]. Prompt removal of galactose/lactose from the diet resolves or prevents these acute symptoms [Walter et al 1999, Sarkar et al 2010] (see Management).
Long-term outcome information for persons with generalized epimerase deficiency galactosemia is limited due to the small number of known affected individuals. Some have demonstrated long-term complications that became evident by early childhood (including sensorineural hearing impairment and physical and cognitive developmental delay and/or learning difficulties) while others have not. Confounding factors include the fact that a majority, but not all, of the individuals reported were born to known consanguineous parents, raising the concern that homozygosity for other autosomal recessive alleles – other than GALE but perhaps genetically linked to GALE – may underlie some of the long-term complications reported. In addition to the features listed in , other rare features reported in survivors include the following:
Normal puberty with no apparent evidence of premature ovarian insufficiency [
Walter et al 1999]
Peripheral Epimerase Deficiency Galactosemia
Neonates with the peripheral form are usually asymptomatic even on a regular milk diet; these infants are only identified following biochemical detection of elevated total galactose on newborn screening.
Children with peripheral epimerase deficiency galactosemia appear to remain asymptomatic even if maintained on a normal milk diet.
Genotype-Phenotype Correlations
Because the numbers of individuals reported with molecularly confirmed epimerase deficiency galactosemia are currently limited, it is difficult to make strong genotype-phenotype correlations. However, some GALE variants have been associated with mild or severe outcomes in multiple affected individuals.
Individuals who have
biallelic GALE alleles that are each associated with higher residual GALE activity in non-peripheral cells, such as (p.Lys257Arg) and (p.Gly319Glu), are more likely to have asymptomatic peripheral epimerase deficiency [
Alano et al 1997,
Openo et al 2006]. However, too few individuals have been described to confirm or refute this prediction.
Nomenclature
Some authors refer to the different forms of galactosemia as type I, type II, type III, and type IV galactosemia, in which:
Prevalence
True prevalence figures are unavailable at this time. Generalized epimerase deficiency galactosemia is very rare; however, epimerase deficiency galactosemia detected by newborn screening may be as frequent as about 1:6,700 among African American infants and about 1:70,000 among US infants of European ancestry [Openo et al 2006, Dias Costa et al 2017].
Differential Diagnosis
GALT deficiency. Galactosemia caused by deficiency of the enzyme galactose-1-phosphate uridylyltransferase (GALT) may be divided into three clinical/biochemical phenotypes: (1) classic galactosemia; (2) clinical variant galactosemia; and (3) Duarte (biochemical variant) galactosemia. This categorization is based on: residual erythrocyte GALT enzyme activity; the levels of galactose metabolites (e.g., erythrocyte galactose-1-phosphate and urine galactitol) that are observed both off and on a lactose-restricted diet; and, most importantly, the likelihood that the affected individual will develop acute and chronic long-term complications. Biallelic pathogenic variants in GALT are causative; inheritance is autosomal recessive.
Classic galactosemia can result in life-threatening complications including feeding issues, failure to thrive, hepatocellular damage, bleeding, and
E coli sepsis in untreated infants. If a lactose-restricted diet is provided during the first ten days of life, the neonatal signs usually quickly resolve and the complications of liver failure, sepsis, and neonatal death are prevented; however, despite adequate treatment from an early age, children with classic galactosemia remain at increased risk for developmental delays, speech issues (termed childhood apraxia of speech and dysarthria), and abnormalities of motor function. The vast majority of women with classic galactosemia manifest premature ovarian insufficiency.
Clinical variant galactosemia can result in life-threatening complications in untreated infants including feeding issues, failure to thrive, hepatocellular damage including cirrhosis, and bleeding. It can occur in individuals of any ancestry with low residual GALT enzyme activity, but is perhaps exemplified by the disease associated with homozygosity for the p.Ser135Leu
GALT allele that occurs at high frequency in native Africans in South Africa, and to a lesser extent in African Americans. Infants with clinical variant galactosemia may be missed if newborn screening only measures blood total galactose level and not erythrocyte GALT enzyme activity, as the hypergalactosemia is not as marked as in classic galactosemia and breath testing is normal. As in classic galactosemia, if a lactose-restricted diet is provided during the first days of life, the severe acute neonatal complications are usually prevented. Long-term outcomes among treated individuals with clinical variant galactosemia may also be milder.
Duarte variant galactosemia (biochemical variant galactosemia). Infants with Duarte variant galactosemia who receive breast milk or a high galactose-containing formula (dairy milk-based formula) are typically asymptomatic and show no greater prevalence of developmental complications than is seen in the general population. Erythrocyte GALT enzyme activity is typically about 25% of control activity. Infants with Duarte variant galactosemia may or may not be detected by newborn screening depending on the GALT enzyme activity cutoff defined by the screening program, but should demonstrate partial deficiency of GALT, potentially with some elevation of galactose metabolites if the baby has consumed breast milk or a dairy milk-based formula).
Galactokinase (GALK) deficiency (OMIM 230200) should be considered in otherwise healthy individuals with cataracts, increased plasma concentration of galactose, and increased urinary excretion of galactitol. Affected individuals have normal GALT enzyme activity and most do not accumulate gal-1P [Hennermann et al 2011] (surprisingly, some individuals with GALK deficiency apparently do accumulate gal-1P) [Rubio-Gozalbo et al 2021]. Affected individuals reported in each of two studies – one including 18 affected individuals identified in Germany [Hennermann et al 2011] and the other describing an independent cohort of 56 affected individuals from 11 countries [Rubio-Gozalbo et al 2021] – displayed a range of acute and long-term outcomes, some of which overlap with complications seen in classic galactosemia. Detection of reduced GALK enzyme activity in hemolysates is diagnostic. Biallelic pathogenic variants in GALK1 are causative; inheritance is autosomal recessive. The prevalence of GALK deficiency in most populations is unknown; however, a study from Germany reported a prevalence of about 1:40,000, which is similar to the prevalence of classic galactosemia in the same population [Hennermann et al 2011]. In other populations the prevalence may be far lower.
Note: Studies in a mouse model confirmed that the cataracts seen in GALK-deficiency, and presumably also in other forms of galactosemia, are caused by accumulation of the galactose metabolite, galactitol, in the lens. Galactitol is an impermeant alcohol which results in increased intracellular osmolality and swelling with loss of plasma membrane redox potential and consequent cell death.
GALM deficiency galactosemia (OMIM 618881) should be considered in individuals who have increased plasma concentration of galactose and may have cataracts, but are otherwise healthy [Timson 2019, Wada et al 2019]. These individuals have a negative Beutler test (ruling out GALT deficiency) and normal GALK and GALE enzyme activities. In affected individuals described to date, galactose-1-phosphate (gal-1P) levels on newborn screening ranged from 0.3 mg/dL to 10.8 mg/dL. Unlike GALE deficiency, the ratio between blood gal-1P and galactose was reported to be normal in GALM galactosemia [Wada et al 2019]. Biallelic pathogenic variants in GALM are causative; identification of biallelic pathogenic variants in GALM on molecular genetic testing is diagnostic (a diagnostic biochemical assay is not available at this time). In one study, the incidence of GALM deficiency was estimated to be almost 1:10,000 in African populations, almost 1:80,000 in the Japanese population, and much lower in many other populations [Iwasawa et al 2019].
Other. A number of other conditions, including the following, can also lead to elevated galactose or galactose metabolites in the blood or urine of an infant consuming milk:
Liver dysfunction / liver failure
Portosystemic venous shunting
Hepatic arteriovenous malformations
Fanconi-Bickel syndrome (OMIM
227810), caused by
biallelic pathogenic variants in
SLC2A2. Individuals with this condition have hepatorenal glycogen accumulation, impaired utilization of glucose and galactose, and proximal tubular nephropathy.
Congenital disorder of glycosylation type 1T (PGM1-CDG, see
Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview), caused by
biallelic pathogenic variants in
PGM1 [
Tegtmeyer et al 2014]. Individuals with this condition can manifest mildly increased galactose-1-phosphate levels in RBC. They may have cleft palate/bifid uvula at birth and abnormal liver enzymes. They can develop liver disease, intermittent hypoglycemia, dilated cardiomyopathy, and exercise intolerance with increased serum creatine kinase.
Management
When epimerase deficiency galactosemia is suspected during the diagnostic evaluation (for example, if total galactose is elevated on newborn screening results), initiation of a galactose/lactose-restricted diet should begin immediately (see Treatment of Manifestations).
To the authors' knowledge, no clinical practice guidelines for epimerase deficiency galactosemia have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with epimerase deficiency galactosemia that is not clearly peripheral, the evaluations summarized in (if not performed as part of the evaluation that led to diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Epimerase Deficiency Galactosemia
View in own window
| System/Concern | Evaluation | Comment |
|---|
|
Constitutional
| Measurement of height, weight, & head circumference | |
|
Neurologic
| Neurologic eval | To incl assessment of tone |
|
Development
| Developmental assessment | To incl motor, adaptive, cognitive, & speech/language eval Eval for early intervention / special education
|
|
Musculoskeletal
| Orthopedics / physical medicine & rehab / PT & OT eval | To incl assessment of:
|
|
Feeding/Nutrition
| Nutrition / feeding team eval | To incl eval of nutritional status & feeding skills |
|
Hepatic
| Liver function tests 1 | |
|
Renal
| Urinalysis 2 | Incl for non-glucose reducing substances Consider renal imaging, such as renal ultrasound, if renal abnormalities are suspected.
|
|
Eyes
| Ophthalmologic eval | To assess for cataracts |
|
Hearing
| Audiologic eval | Assess for sensorineural hearing loss. |
Genetic
counseling
| By genetics professionals 3 | To inform affected persons & their families re nature, MOI, & implications of epimerase deficiency galactosemia in order to facilitate medical & personal decision making |
Family support
& resources
| | Assess need for:
|
- 1.
To include serum AST, ALT, albumin, total protein, total and conjugated bilirubin, prothrombin time, and partial thromboplastin time
- 2.
One affected person who died in the neonatal period was reported to have large kidneys with intratubular renal calcifications [Dias Costa et al 2017].
- 3.
Medical geneticist, certified genetic counselor, certified advanced genetic nurse
Treatment of Manifestations
Generalized epimerase deficiency galactosemia. The acute and potentially lethal symptoms of generalized epimerase deficiency galactosemia are prevented or corrected by a galactose/lactose-restricted diet (see ).
Intermediate epimerase deficiency galactosemia. Individuals with intermediate epimerase deficiency galactosemia are typically treated with dietary galactose/lactose restriction, at least in infancy (see ). They may be at an (as-yet unknown) increased risk for long-term complications including learning impairment and/or cataracts. Continued breastfeeding or exposure to a milk-based formula containing high levels of galactose/lactose may therefore be inadvisable for these infants; however, insufficient data exist to make firm recommendations.
Table 4.
Treatment of Manifestations in Individuals with Generalized or Intermediate Epimerase Deficiency Galactosemia
View in own window
Principle/
Manifestation/
Concern | Treatment | Considerations/Other |
|---|
Galactose/lactose-
restricted diet 1
| In infants: switch from breast milk or a milk-based formula to a formula w/trace levels of galactose or lactose (e.g., soy formula). |
|
| In older children: Dietary restriction involves continued restriction of dairy products. | While dietary restriction of high-galactose dairy products is recommended, a diet that restricts non-dairy sources of galactose (legumes, some fruits & vegetables, organ meat) is NOT recommended. 3 |
Developmental
delay
| See Developmental Delay / Intellectual Disability Management Issues. | |
Contractures &
clubfoot
| Standard treatment per orthopedist | |
Poor weight gain /
Failure to thrive
| Feeding therapy | |
|
Cataracts
| Mature cataracts that do not resolve w/dietary restriction of galactose/lactose may require surgical removal. | |
|
Hearing loss
| Hearing aids may be helpful; per otolaryngologist. | Community hearing services through early intervention or school district |
Family/
Community
| Ensure appropriate social work involvement to connect families w/local resources & support. | Consider involvement in adaptive sports or Special Olympics. |
- 1.
Restriction of dietary galactose/lactose appears to correct or prevent the common acute signs and symptoms of the disorder: hepatic dysfunction, renal dysfunction, and mild cataracts. However, it may not correct tissue damage that occurred due to prolonged galactose exposure (e.g., hepatic cirrhosis or mature cataracts) (see Prevention of Primary Manifestations).
- 2.
Elemental formula should not be prescribed for infants with generalized epimerase deficiency galactosemia because the GALE enzyme is required for the endogenous biosynthesis of UDP-galactose; that is, persons with epimerase deficiency galactosemia may require trace environmental sources of galactose.
- 3.
Historically, some healthcare providers recommended that individuals with classic galactosemia also abstain from non-dairy foods that contain more than trace levels of galactose/lactose; however, most non-dairy foods have been deemed acceptable for individuals with classic galactosemia [Van Calcar et al 2014].
Peripheral epimerase deficiency galactosemia. Individuals with peripheral epimerase deficiency galactosemia do not require any dietary restriction.
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
IEP services:
An IEP provides specially designed instruction and related services to children who qualify.
IEP services will be reviewed annually to determine whether any changes are needed.
Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction. Physical therapy is recommended to maximize mobility.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Communication issues. Affected individuals with speech and language delay typically require speech therapy until the complication has been resolved.
Prevention of Primary Manifestations
The challenge in treating an asymptomatic newborn with epimerase deficiency galactosemia is that it may take months to obtain the results of tests used to distinguish peripheral epimerase deficiency galactosemia from intermediate epimerase deficiency galactosemia (see Establishing the Diagnosis, Additional Testing); furthermore, such tests may not be available. The most conservative approach, therefore, is to advise dietary restriction of galactose/lactose for all infants with epimerase deficiency galactosemia, relaxing the restriction as warranted once a more accurate diagnosis has been confirmed.
In generalized epimerase deficiency galactosemia restriction of dietary galactose/lactose appears to correct or prevent the common acute signs and symptoms of the disorder: hepatic dysfunction, renal dysfunction, and mild cataracts. Presumably, as in classic galactosemia, dietary treatment would not correct profound tissue damage resulting from prolonged galactose exposure (e.g., hepatic cirrhosis or mature cataracts) or structural defects that likely originated in utero (e.g., cardiomyopathy).
In generalized epimerase deficiency galactosemia dietary restriction of galactose/lactose also prevents early feeding issues, vomiting, poor weight gain, hepatic dysfunction, and cataracts.
Surveillance
Table 5.
Recommended Surveillance for Individuals with Generalized or Intermediate Epimerase Deficiency Galactosemia
View in own window
| System/Concern | Evaluation | Frequency |
|---|
|
Constitutional
| Measurement of growth parameters | At each visit |
|
Biochemical
| Assessment of hemolysate gal-1P or urinary galactitol 1, 2 | At each visit or as indicated by any ongoing concerns or relevant dietary changes |
|
Development
| Monitor developmental progress & educational needs. | At each visit |
|
Musculoskeletal
| Physical medicine, OT/PT assessment |
|
Eyes
| Ophthalmology eval | As clinically indicated (assuming the affected person is on a galactose-restricted diet) |
|
Hearing
| Audiology eval | At least annually in infancy & childhood or as clinically indicated |
Family/
Community
| Assess family need for social work support (e.g., palliative/respite care, home nursing, other local resources) & care coordination. | At each visit |
OT = occupational therapy; PT = physical therapy
- 1.
Especially if the diet is to be normalized
- 2.
Acceptable levels of gal-1P in GALE deficiency are not known but are estimated from experience with classic galactosemia to be <3.5 mg/100 mL in red blood cells.
Agents/Circumstances to Avoid
Persons with generalized epimerase deficiency galactosemia should be on a galactose/lactose-restricted diet, certainly as infants and perhaps for life.
Persons with intermediate epimerase deficiency galactosemia may be placed on a galactose/lactose-restricted diet, either transiently or long term. Assessment of hemolysate gal-1P and/or urinary galactitol following a galactose challenge (e.g., 2 weeks on a normal diet) may help determine if an individual should remain on a galactose/lactose-restricted diet for longer periods of time.
Evaluation of Relatives at Risk
If prenatal testing has not been performed (see Genetic Counseling), each at-risk newborn sib should be treated with dietary restriction of galactose from birth until results of diagnostic testing are available. Diagnostic evaluations can include the following:
Note: If there are concerns about the reliability of the prenatal testing, soy-based formula may be given while the diagnostic testing is being performed.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Epimerase deficiency galactosemia is inherited in an autosomal recessive manner.
Carrier Detection
Molecular genetic testing. Carrier testing for at-risk relatives requires prior identification of the GALE pathogenic variants in the family.
Note: Although biochemical testing to detect carriers is also a possibility, the ranges for control and carrier GALE enzyme activity overlap, thus making molecular genetic testing the preferred method for carrier detection.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the GALE pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Note: Theoretically, prenatal testing could be accomplished by enzymatic studies of amniocytes or CVS tissue; however, due to lack of a GALE reference range for the relevant sample type and appropriate controls, this testing is not typically offered on a clinical basis.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
British Inherited Metabolic Disease Group (BIMDG)
TEMPLE (Tools Enabling Metabolic Parents LEarning)
United Kingdom
Medical Home Portal
The Galactosemia Foundation
350 Northern Boulevard
Suite 324 - 1079
Albany NY 12204-1000
Phone: 866-900-7421
Email: outreach@galactosemia.org
Metabolic Support UK
United Kingdom
Phone: 0845 241 2173
Newborn Screening in Your State
Health Resources & Services Administration
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Epimerase Deficiency Galactosemia: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
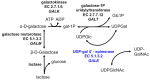
Galactose is metabolized in humans and other species by the three-enzyme Leloir pathway comprising the enzymes galactokinase (GALK, EC 2.7.1.6), galactose 1-P uridylyltransferase (GALT, EC 2.7.7.12), and UDP-galactose 4'-epimerase (GALE, EC 5.1.3.2). A fourth enzyme, galactose mutarotase (GALM, EC 5.1.3.3), catalyzes the epimerization of β-D-galactose, released from lactose, to α-D-galactose, which is a substrate for GALK. As illustrated in , GALE catalyzes an essential step in this pathway converting UDP-galactose to UDP-glucose. GALE is a reversible enzyme that also catalyzes the synthesis of UDP-galactose from UDP-glucose when other sources of UDP-galactose are limiting. Functioning outside of the Leloir pathway, GALE also interconverts UDP-N-acetyl galactosamine and UDP-N-acetylglucosamine. All four of these UDP-sugars are essential substrates for the biosynthesis of glycoproteins and glycolipids in humans.
As in classic galactosemia, the cataracts associated with epimerase deficiency galactosemia are believed to be caused by galactitol accumulation in the ocular lens; it is possible, but not proven, that other acute findings may be caused by tissue accumulation of gal-1P (galactose-1-phosphate) or other metabolites.
Persons with epimerase deficiency galactosemia who are exposed to galactose demonstrate abnormal accumulation of UDP-galactose (UDP-gal). However, because GALE is required in humans for the endogenous biosynthesis of UDP-gal and also UDP-N-acetylgalactosamine (UDP-galNAc), at least part of the pathophysiology of epimerase deficiency galactosemia may result from inadequate production of these compounds, especially in utero, ostensibly leading to deficient or aberrant production of glycoproteins and glycolipids including cerebrosides.
Mechanism of disease causation. Loss of function
No individuals with complete loss of GALE enzyme activity in non-peripheral cells have been reported [Kalckar 1965]. In addition, fruit fly [Sanders et al 2010, Daenzer et al 2012] and C elegans [Brokate-Llanos et al 2014] models for GALE impairment suggest that complete loss of GALE enzyme activity may be lethal.
The c.280G>A (p.Val94Met) pathogenic variant, which is associated with a severe presentation, leaves approximately 5% residual enzyme activity with regard to UDP-gal metabolism and close to 25% residual enzyme activity with regard to UDP-galNAc metabolism [Wohlers et al 1999, Wohlers & Fridovich-Keil 2000].
Table 6.
Notable GALE Pathogenic Variants
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Chapter Notes
Acknowledgments
The authors gratefully acknowledge the time and efforts of the many patients, families, health care professionals, and scientists who have brought our knowledge of epimerase deficiency galactosemia to its current state. JLFK also gratefully acknowledges funding from the National Institutes of Health and the Galactosemia Foundation.
Revision History
4 March 2021 (ma) Comprehensive update posted live
16 June 2016 (ma) Comprehensive update posted live
24 October 2013 (me) Comprehensive update posted live
25 January 2011 (me) Review posted live
31 August 2010 (jfk) Original submission
References
Literature Cited
Alano A, Almashanu S, Chinsky JM, Costeas P, Blitzer MG, Wulfsberg EA, Cowan TM. Molecular characterization of a unique patient with epimerase-deficiency galactosaemia.
J Inherit Metab Dis. 1998;21:341–50. [
PubMed: 9700591]
Alano A, Almashanu S, Maceratesi P, Reichardt J, Panny S, Cowan TM. UDP-galactose-4-epimerase deficiency among African-Americans: evidence for multiple alleles. J Invest Med. 1997;45:191A.
Berry GT, Walter JH, Fridovich-Keil JL. Disorders of galactose metabolism. In: Saudubray J-M, Baumgartner M, Garcia-Cazorla A, Walter JH, eds. Inborn Metabolic Diseases: Diagnosis and Treatment. Ch 6. 7 ed. Springer-Verlag. 2020.
Chen J, Meyers GA, Bennett MJ. An interference-free two-step enzyme assay with UPLC-tandem mass spectrometric product measurement for the clinical diagnosis of uridine diphosphate galactose-4-epimerase deficiency.
J Chromatogr B Analyt Technol Biomed Life Sci. 2014;2014;959:5–9. [
PubMed: 24732214]
Daenzer JM, Sanders RD, Hang D, Fridovich-Keil JL. UDP-galactose 4'-epimerase activities toward UDP-Gal and UDP-GalNAc play different roles in the development of Drosophila melanogaster.
PLoS Genet. 2012;8:e1002721. [
PMC free article: PMC3359975] [
PubMed: 22654673]
Dias Costa F, Ferdinandusse S, Pinto C, Dias A, Keldermans L, Quelhas D, Matthijs G, Mooijer PA, Diogo L, Jaeken J, Garcia P. Galactose epimerase deficiency: expanding the phenotype.
JIMD Rep. 2017;37:19–25. [
PMC free article: PMC5740041] [
PubMed: 28247339]
Garibaldi LR, Canini S, Superti-Furga A, Lamedica G, Filocamo M, Marchese N, Borrone C. Galactosemia caused by generalized uridine diphosphate galactose-4-epimerase deficiency.
J Pediatr. 1983;103:927–30. [
PubMed: 6549612]
Gitzelmann R. Deficiency of uridine diphosphate galactose 4-epimerase in blood cells of an apparently healthy infant. Preliminary communication.
Helv Paediatr Acta. 1972;27:125–30. [
PubMed: 4644860]
Henderson MJ, Holton JB, MacFaul R. Further observations in a case of uridine diphosphate galactose-4-epimerase deficiency with a severe clinical presentation.
J Inherit Metab Dis. 1983;6:17–20. [
PubMed: 6408303]
Hennermann JB, Schadewaldt P, Vetter B, Shin YS, Mönch E, Klein J. Features and outcome of galactokinase deficiency in children diagnosed by newborn screening.
J Inherit Metab Dis. 2011;34:399–407. [
PubMed: 21290184]
Holton JB, Gillett MG, MacFaul R, Young R. Galactosaemia: a new severe variant due to uridine diphosphate galactose-4-epimerase deficiency.
Arch Dis Child. 1981;56:885–7. [
PMC free article: PMC1627389] [
PubMed: 7305435]
Iwasawa S, Kikuchi A, Wada Y, Arai-Ichinoi N, Sakamoto O, Tamiya G, Kure S. The prevalence of GALM mutations that cause galactosemia: A database of functionally evaluated variants.
Mol Genet Metab. 2019;126:362–7. [
PubMed: 30910422]
Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland.
Nature. 2017;549:519–22. [
PubMed: 28959963]
Kalckar HM. Galactose metabolism and cell "sociology".
Science. 1965;150:305–13. [
PubMed: 5319435]
Openo KK, Schulz JM, Vargas CA, Orton CS, Epstein MP, Schnur RE, Scaglia F, Berry GT, Gottesman GS, Ficicioglu C, Slonim AE, Schroer RJ, Yu C, Rangel VE, Keenan J, Lamance K, Fridovich-Keil JL. Epimerase-deficiency galactosemia is not a binary condition.
Am J Hum Genet. 2006;78:89–102. [
PMC free article: PMC1380226] [
PubMed: 16385452]
Park HD, Park KU, Kim JQ, Shin CH, Yang SW, Lee DH, Song YH, Song J. The molecular basis of UDP-galactose-4-epimerase (GALE) deficiency galactosemia in Korean patients.
Genet Med. 2005;7:646–9. [
PubMed: 16301867]
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med. 2015;17:405–24. [
PMC free article: PMC4544753] [
PubMed: 25741868]
Rubio-Gozalbo ME, Derks B, Das AM, Meyer U, Möslinger D, Couce ML, Empain A, Ficicioglu C, Juliá Palacios N, De Los Santos De Pelegrin MM, Rivera IA, Scholl-Bürgi S, Bosch AM, Cassiman D, Demirbas D, Gautschi M, Knerr I, Labrune P, Skouma A, Verloo P, Wortmann SB, Treacy EP, Timson DJ, Berry GT. Galactokinase deficiency: lessons from the GalNet registry.
Genet Med. 2021;23:202–10. [
PMC free article: PMC7790741] [
PubMed: 32807972]
Sanders RD, Sefton JM, Moberg KH, Fridovich-Keil JL. UDP-galactose 4' epimerase (GALE) is essential for development of Drosophila melanogaster.
Dis Model Mech. 2010;3:628–38. [
PMC free article: PMC2931539] [
PubMed: 20519568]
Sardharwalla IB, Wraith JE, Bridge C, Fowler B, Roberts SA. A patient with severe type of epimerase deficiency galactosaemia.
J Inherit Metab Dis. 1988;11:249–51. [
PubMed: 3141714]
Sarkar M, Bose SS, Mondal G, Chatterjee S. Generalized epimerase deficiency galactosemia.
Indian J Pediatr. 2010;77:909–10. [
PubMed: 20725869]
Seo A, Gulsuner S, Pierce S, Ben-Harosh M, Shalev H, Walsh T, Krasnov T, Dgany O, Doulatov S, Tamary H, Shimamura A, King MC. Inherited thrombocytopenia associated with mutation of UDP-galactose-4-epimerase (GALE).
Hum Mol Genet. 2019;28:133–42. [
PMC free article: PMC6298239] [
PubMed: 30247636]
Tegtmeyer LC, Rust S, van Scherpenzeel M, Ng BG, Losfeld ME, Timal S, Raymond K, He P, Ichikawa M, Veltman J, Huijben K, Shin YS, Sharma V, Adamowicz M, Lammens M, Reunert J, Witten A, Schrapers E, Matthijs G, Jaeken J, Rymen D, Stojkovic T, Laforêt P, Petit F, Aumaître O, Czarnowska E, Piraud M, Podskarbi T, Stanley CA, Matalon R, Burda P, Seyyedi S, Debus V, Socha P, Sykut-Cegielska J, van Spronsen F, de Meirleir L, Vajro P, DeClue T, Ficicioglu C, Wada Y, Wevers RA, Vanderschaeghe D, Callewaert N, Fingerhut R, van Schaftingen E, Freeze HH, Morava E, Lefeber DJ, Marquardt T. Multiple phenotypes in phosphoglucomutase 1 deficiency.
N Engl J Med. 2014;370:533–42. [
PMC free article: PMC4373661] [
PubMed: 24499211]
Van Calcar SC, Bernstein LE, Rohr FJ, Scaman CH, Yannicelli S, Berry GT. A re-evaluation of life-long severe galactose restriction for the nutrition management of classic galactosemia.
Mol Genet Metab. 2014;112:191–7. [
PubMed: 24857409]
Wada Y, Kikuchi A, Arai-Ichinoi N, Sakamoto O, Takezawa Y, Iwasawa S, Niihori T, Nyuzuki H, Nakajima Y, Ogawa E, Ishige M, Hirai H, Sasai H, Fujiki R, Shirota M, Funayama R, Yamamoto M, Ito T, Ohara O, Nakayama K, Aoki Y, Koshiba S, Fukao T, Kure S. Biallelic GALM pathogenic variants cause a novel type of galactosemia.
Genet Med. 2019;21:1286–94. [
PubMed: 30451973]
Walter JH, Roberts RE, Besley GT, Wraith JE, Cleary MA, Holton JB, MacFaul R. Generalised uridine diphosphate galactose-4-epimerase deficiency.
Arch Dis Child. 1999;80:374–6. [
PMC free article: PMC1717903] [
PubMed: 10086948]
Wohlers TM, Christacos NC, Harreman MT, Fridovich-Keil JL. Identification and characterization of a mutation, in the human UDP-galactose-4-epimerase gene, associated with generalized epimerase-deficiency galactosemia.
Am J Hum Genet. 1999;64:462–70. [
PMC free article: PMC1377755] [
PubMed: 9973283]
Wohlers TM, Fridovich-Keil JL. Studies of the V94M-substituted human UDPgalactose-4-epimerase enzyme associated with generalized epimerase-deficiency galactosaemia.
J Inherit Metab Dis. 2000;23:713–29. [
PubMed: 11117433]