Clinical Description
Huppke-Brendel syndrome (HBS) is characterized by cataract, sensorineural deafness, and severe developmental delay in all reported individuals. To date, six individuals with HBS have been reported in the literature [Horváth et al 2005, Huppke et al 2012, Chiplunkar et al 2016].
Ocular features. Bilateral congenital cataracts were reported in all affected individuals. Affected individuals presented with poor visual fixation and rotary nystagmus. Two individuals underwent cataract extraction in early infancy; there was improvement in visual fixation and nystagmus in one child [Horváth et al 2005] and no improvement in vision in the other [Chiplunkar et al 2016].
Sensorineural hearing loss manifests during infancy. Brain stem auditory evoked potentials in two individuals showed absent waveforms. Otoacoustic emissions were absent bilaterally in one individual.
Neurologic features. Axial hypotonia was present in all infants. Motor delay was apparent in the first few months of life in all reported individuals. Lack of head control and paucity of movements in the limbs were evident. None of the individuals reported to date were able to sit or walk independently. None learned to speak. Developmental progress has been reported only in one individual who received copper histidinate therapy from age five months [Horváth et al 2005]. Follow up at age 13 months in this individual showed good head control, rolling over, reaching out for objects, and improved alertness and communication.
Deep tendon reflexes were normal and symmetric.
Two of the six reported individuals had seizures.
Orthopedic complications. Affected individuals are at increased risk for scoliosis and joint contractures [Author, personal communication].
Other. Hypopigmented hair and hypogenitalism were reported in one individual [Chiplunkar et al 2016]. This child had micropenis with bilaterally descended testes. The hair was uniformly hypopigmented and sparse. Hair analysis under polarized light microscopy showed uniform and finely granulated melanin pigment and no clumps. There was no kinking or abnormal polarization.
Prognosis. All affected individuals died between age ten months and six years. Causes of death included pneumonia, kidney failure, and multiorgan failure. The cause of multiorgan failure was not reported.
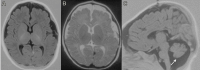
Neuroimaging. Brain MRI showed hypomyelination, cerebellar hypoplasia mainly affecting the vermis, hypoplasia of the temporal lobes, and wide subarachnoid spaces (see ) [Horváth et al 2005, Huppke et al 2012, Chiplunkar et al 2016].
Brain MRI in a child with HBS at age four months A, B. T1-weighted axial view (A) and T2-weighted axial view (B) show presence of myelin only in the posterior limb of the internal capsule, indicating delayed myelination. Widened subarachnoid spaces are (more...)
Histopathology on muscle biopsy
Subsarcolemmal proliferation and vacuolization in few type 1 fibers are reported. No typical ragged red fibers or ragged blue fibers or COX-negative fibers are seen [
Horváth et al 2005,
Chiplunkar et al 2016].
Biochemical measurements of the respiratory chain enzymes showed significantly reduced activity of COX with 30% residual activity in one individual [
Horváth et al 2005] and 35% residual activity in another [
Chiplunkar et al 2016].