Summary
Clinical characteristics.
INSR-related severe insulin resistance syndrome (INSR-SIRS) comprises a phenotypic spectrum that is a continuum from the severe phenotype of Donohue syndrome to the milder phenotype of Rabson-Mendenhall syndrome (RMS).
Donohue syndrome is characterized by severe insulin resistance (hyperinsulinemia with associated fasting hypoglycemia and postprandial hyperglycemia), severe prenatal growth restriction, postnatal growth failure, hypotonia, developmental delay, characteristic facies (proptosis, infraorbital folds, large, low-set, posteriorly rotated ears, thick vermilion of the upper and lower lips, and gingival hypertrophy), and organomegaly involving the heart, kidneys, liver, spleen, and ovaries. Death usually occurs before age one year.
RMS, at the milder end of the spectrum, is characterized by severe insulin resistance that, although not as severe as that of Donohue syndrome, is nonetheless accompanied by fluctuations in blood glucose levels, diabetic ketoacidosis, and – in the second decade – microvascular complications. Findings can range from severe growth delay and intellectual disability to normal growth and development. Facial features can be milder than those of Donohue syndrome. Complications of longstanding hyperglycemia are the most common cause of death. While death usually occurs in the second decade, some affected individuals live longer.
Diagnosis/testing.
The diagnosis of INSR-SIRS is established in a proband with characteristic clinical, laboratory, radiographic, and prenatal ultrasound findings and biallelic INSR pathogenic variants identified by molecular genetic testing.
Management.
Treatment of manifestations: Donohue syndrome: no effective treatments for insulin resistance or other manifestations of Donohue syndrome are currently available. Frequent feedings as well as increased protein content of evening feedings can help prevent fasting hypoglycemia.
RMS: Insulin sensitizers are used first to decrease levels of glucose and glycosylated hemoglobin (HbA1c); however, their effect diminishes with time, often requiring dose adjustments and multidrug therapy. When hyperglycemia persists, insulin is started – usually in high doses, especially during the treatment of diabetic ketoacidosis. Standard treatment for hypothyroidism; oral contraceptives, antiandrogen therapies, and gonadotropin-releasing hormone agonists can be used to treat hyperandrogenism; oophorectomy may be needed for enlarged ovaries; nutritional, developmental, and educational support; rigorous workup and treatment of intercurrent infections; beta-blockers for cardiomyopathy; treatment of nephrocalcinosis per nephrologist; treatment of cholestasis per gastroenterologist; treatment of rectal prolapse per surgeon; standard treatments for malignancies; social work and family support.
Surveillance: Capillary glucose levels during fasting and after feeding, when clinically indicated, or continuous monitoring; HbA1c, insulin, and C-peptide levels every three months; thyroid function and hyperandrogenism lab assessment every six months or as clinically indicated; ovarian ultrasound every three months until age two years, then every six months or as indicated; nutrition and growth assessment at each visit; assess psychomotor development every three months; assess for recurrent infections at each visit; echocardiogram and cardiac MRI every six months (each test alternating every three months) until age two years, then annually (each test alternating every six months) or as indicated; urine calcium and kidney ultrasound every six months; gynecologic evaluation for endometrial cancer in those with abnormal vaginal bleeding.
Agents/circumstances to avoid: In Donohue syndrome, avoid agents that cause hypoglycemia, prolonged fasting, and contact with persons with contagious disease. In RMS avoid agents that cause hypoglycemia, high-carbohydrate diet, and contact with persons with contagious disease.
Genetic counseling.
INSR-SIRS is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an INSR pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting biallelic pathogenic variants and being affected with INSR-SIRS, a 50% chance of inheriting one pathogenic variant and being heterozygous, and a 25% chance of inheriting neither of the familial INSR pathogenic variants. Heterozygotes are usually asymptomatic but may have features of the allelic disorder type A insulin resistance. Once the INSR pathogenic variants have been identified in an affected family member, heterozygote testing for at-risk relatives and prenatal and preimplantation genetic testing are possible. (Of note: Heterozygotes for an INSR pathogenic variant are at increased risk for gestational diabetes and require monitoring for glucose intolerance before and during pregnancy.)
GeneReview Scope
Table
Donohue syndrome 1 Rabson-Mendenhall syndrome
Diagnosis
Suggestive Findings
INSR-related severe insulin resistance syndrome (INSR-SIRS) should be suspected in individuals with the following clinical, laboratory, and imaging findings of Donohue syndrome or Rabson-Mendenhall syndrome (RMS).
Clinical Findings
Donohue syndrome
- Progressive intrauterine growth restriction (IUGR) from the early third trimester and postnatal poor weight gain, growth deficiency, and reduced subcutaneous fat
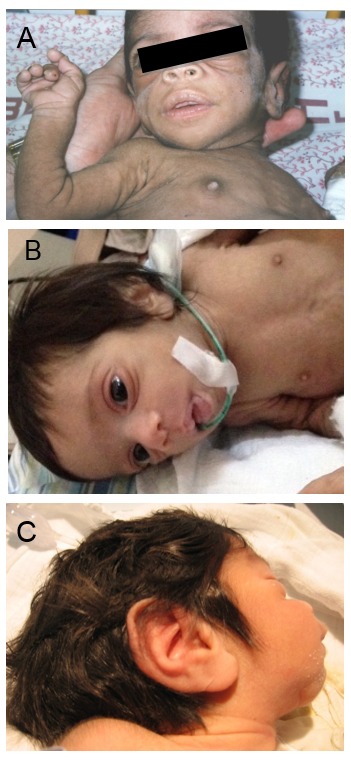
- Dysmorphic facial features (Figure 1) characterized by proptosis, infraorbital folds, large, low-set, posteriorly rotated ears, thick vermilion of the upper and lower lips, and gingival hypertrophy [Grasso et al 2013]
- Hypotonia
- Developmental delay
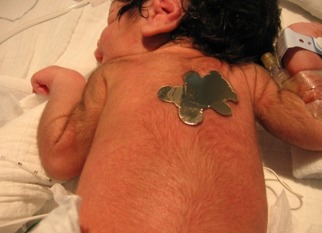
- Integument, including dry skin, hypertrichosis (Figure 2), and acanthosis nigricans
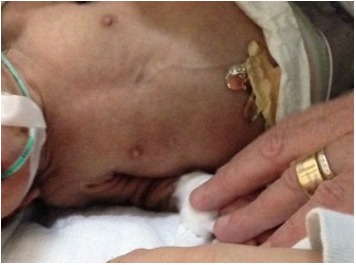
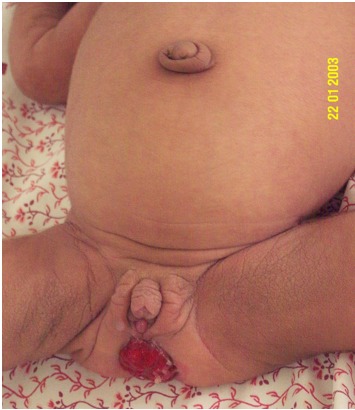
- Prominent nipples (Figure 3)
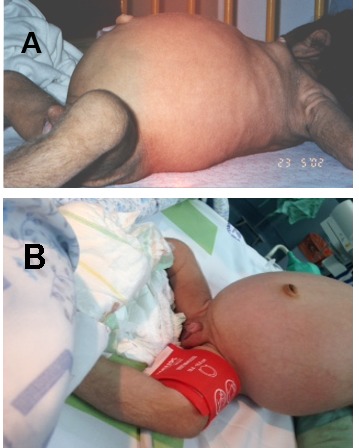
- Abdominal distention (Figure 4) and organomegaly
- Genital enlargement (males and females) (Figure 4B)
- Rectal hypertrophy and prolapse (Figure 5)
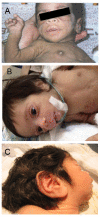
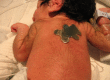
Figure 2.
Hypertrichosis is a feature in all individuals with Donohue syndrome. Reproduced from Falik Zaccai et al [2014]
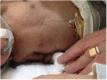
Figure 3.
Prominent nipples are a typical finding in neonates with Donohue syndrome.
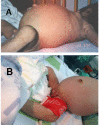
Figure 4
A and B. Abdominal distention B. Labial hypertrophy and clitoromegaly in a girl age four months with Donohue syndrome
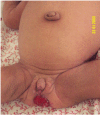
Figure 5.
Rectal hypertrophy and prolapse in an infant with Donohue syndrome.
RMS
- Dysmorphic facial features that can resemble those of Donohue syndrome [Ben Abdelaziz et al 2016] or can be milder [Musso et al 2004]
- Integument, including hypertrichosis and acanthosis nigricans
- Prominent nipples
- Genital enlargement (males and females)
- Early dental eruption and dental crowding [Bathi et al 2010]
- Growth deficiency (less severe than in Donohue syndrome) [Tuthill et al 2007]
- Developmental delay (in some individuals) [Ben Abdelaziz et al 2016, Sinnarajah et al 2016]
Laboratory Findings
Donohue syndrome
- Severe hyperinsulinemia. Extremely high plasma insulin and C-peptide levels with fluctuating blood glucose levels (typically fasting hypoglycemia and postprandial hyperglycemia)
- Ketoacidosis not reported.
RMS
- Birth to age one year. Severe hyperinsulinemia, with extremely high plasma insulin and C-peptide levels with fluctuating blood glucose levels (typically fasting hypoglycemia and postprandial hyperglycemia)
- Age one year and older. Insulin levels decline steadily, initially resulting in increased glucose levels and fewer hypoglycemic events, and subsequently resulting in increased risk for ketoacidosis.
- Low triglyceride levels, high high-density lipoprotein (HDL) levels [Semple et al 2009]
- High adiponectin levels [Semple et al 2008]
Imaging Findings
Donohue syndrome and RMS
- Hypertrophic cardiomyopathy
- Enlarged kidneys, liver, and spleen
- Nephrocalcinosis
- Enlarged polycystic ovaries
Establishing the Diagnosis
The diagnosis of INSR-SIRS is established in a proband with suggestive findings and biallelic INSR pathogenic (or likely pathogenic) variants identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of biallelic INSR variants of uncertain significance (or of one known INSR pathogenic variant and one INSR variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing). Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1), whereas comprehensive genomic testing does not (see Option 2).
Option 1
Single-gene testing. Sequence analysis of INSR is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
A multigene panel that includes INSR and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. Of note, given the rarity of INSR-SIRS, some panels for diabetes mellitus may not include this gene. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in INSR-Related Severe Insulin Resistance Syndrome
Clinical Characteristics
Clinical Description
INSR-related severe insulin resistance syndrome (INSR-SIRS) comprises a phenotypic continuum from the severe phenotype of Donohue syndrome to the milder phenotype of Rabson-Mendenhall syndrome (RMS). To date, fewer than 100 individuals have been identified with INSR-SIRS [Termote et al 2016]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
INSR-Related Severe Insulin Resistance Syndrome: Comparison of Phenotypes by Select Features
Donohue Syndrome
Donohue syndrome is characterized by severe insulin resistance, growth failure, hypotonia, developmental delay, characteristic facies, and organomegaly.
Endocrine manifestations. Insulin resistance with hyperinsulinemia (fasting hypoglycemia and postprandial hyperglycemia) is present from birth. There are currently no effective treatments for the insulin resistance. Postprandial hyperglycemia does not respond to insulin treatment [Semple et al 2010] or glucose-lowering therapies such as metformin [Musso et al 2004].
Enlargement of the external genitalia occurs in both males (enlargement of the penis) and females (labial hypertrophy and clitoral enlargement). Ovarian enlargement is characteristic; ovarian ultrasonography usually reveals multiple peripheral cysts, as seen in idiopathic polycystic ovary syndrome. Cysts may become very large and vulnerable to hemorrhage or torsion, and surgical removal may be required [Semple et al 2011]. Prominent nipples are seen in all individuals.
Central hypothyroidism has been reported [Baqir et al 2012, Falik Zaccai et al 2014].
Growth deficiency. Birth weight, length, and head circumference are below the third centile. However, weight and/or length are more affected than head circumference [Dagdeviren Cakir et al 2020]. Neonates continue to have poor weight gain and severe growth deficiency; the average weight at age one year is 4-5 kg [Longo et al 2002]. Reduced subcutaneous fat is present in all individuals.
Developmental delay / intellectual disability. Most infants have severe global developmental delay, including speech and motor as well as cognitive impairment [Falik Zaccai et al 2014]. Most individuals do not reach any significant developmental milestones prior to early demise [Falik Zaccai et al 2014, Joshi et al 2021]. Axial hypotonia and muscle atrophy are also observed [Baqir et al 2012]. Intellectual disability is thought to be a consequence of recurrent, severe hypoglycemic episodes [Ben Abdelaziz et al 2016].
Characteristic facies include proptosis, infraorbital folds, large, low-set, posteriorly rotated ears, thick vermilion of the upper and lower lips, and gingival hypertrophy [Grasso et al 2013] (see Figure 1). Macroglossia has been reported but may be secondary to prolonged therapy with insulin-like growth factor 1 (IGF-1) [Joshi et al 2021].
Recurrent infections. Recurrent bacterial infections including pneumonia and urinary tract infection are a common cause of death; health care-acquired infections may contribute to recurrent infections [Rojek et al 2023].
Integument. Hypertrichosis and hyperkeratosis are present at birth in all affected infants. Acanthosis nigricans can be apparent at birth or in early infancy [Musso et al 2004].
Cardiac manifestations. Hypertrophic cardiomyopathy is present in 30% of infants and is a major cause of death [Geffner et al 1987, Hovnik et al 2013, Falik Zaccai et al 2014].
Renal manifestations. Enlarged kidneys are common. Abnormalities of the renin-aldosterone system are reported, resulting in hypokalemia, hyperaldosteronism, and hyperreninemia [Grasso et al 2013]. Hypercalciuria and nephrocalcinosis are often seen during the first months of life and in some instances can be detected prenatally [Simpkin et al 2014]. Kidney function as measured by glomerular filtration rate and plasma creatinine concentration is normal [Grasso et al 2013, Simpkin et al 2014].
Liver/gastrointestinal manifestations. Cholestasis is common in the neonatal period [Hovnik et al 2013, Kawashima et al 2013]. Hepatomegaly occurs without liver dysfunction. Rectal prolapse or hypertrophy can be present, which sometimes requires colostomy [Weber et al 2014].
Dental findings include macrodontia and dental crowding.
Prognosis. Death usually occurs during the first year of life. The major causes of death are complications of hypoglycemia, recurrent bacterial infections [Elders et al 1982, de Bock et al 2012], and cardiomyopathy [Grasso et al 2013].
Rabson-Mendenhall Syndrome (RMS)
RMS, at the milder end of the spectrum, is characterized by severe insulin resistance with fluctuations in blood glucose levels, diabetic ketoacidosis, and microvascular complications. Findings can range from severe growth deficiency and intellectual disability to normal growth and development. Facial features can be milder than those of Donohue syndrome.
Endocrine manifestations. Insulin resistance with hyperinsulinemia is less severe than that of Donohue syndrome. Fluctuations in blood glucose levels occur with fasting hypoglycemia and postprandial hyperglycemia from birth. After age one year, insulin levels decline steadily, initially resulting in increased glucose levels and fewer hypoglycemic events, and subsequently resulting in increased risk for ketoacidosis. Morbidity in older individuals with RMS results from microvascular complications of prolonged hyperglycemia and hyperinsulinemia (e.g., proliferative retinopathy, peripheral neuropathy, and renal vascular complications) [Musso et al 2004, Carrasco de la Fuente et al 2010, Jiang et al 2011].
Enlargement of the external genitalia in both males and females typically appears later in childhood in individuals with RMS compared to those with Donohue syndrome. Enlarged ovaries are reported in some individuals and may be complicated by multiple cysts and development of tumors [Parker & Semple 2013]. Prominent nipples are also seen.
Thyroid abnormalities reported in RMS include high prevalence of thyroid nodules and thyromegaly [Kushchayeva et al 2019].
Growth is variable in individuals with RMS, from severe growth deficiency that includes reduction of weight, length, and head circumference [Dagdeviren Cakir et al 2020] to normal growth [Musso et al 2004]. In those with growth deficiency, linear growth does not improve with human growth hormone treatment even when the child's growth hormone levels are low [Musso et al 2004, Kim et al 2012, Brown et al 2013].
Developmental delay / intellectual disability. Developmental delay can range from severe to normal development [de Kerdanet et al 2015, Ben Abdelaziz et al 2016]. More than 50% of individuals with RMS are reported to have mild intellectual disability [Ben Abdelaziz et al 2016, Angelidi et al 2021].
Dysmorphic facial features in individuals with RMS can be more subtle than facial features characteristic of Donohue syndrome (e.g., proptosis, infraorbital folds, large, low-set, posteriorly rotated ears, thick vermilion of the upper and lower lips, and gingival hypertrophy). The face can show increased vertical growth, increased gonial angle, macroglossia, macrodontia, dental crowding, dental prematurity, and dysplastic dentition [Joshi et al 2021]. Early dental eruption, dental crowding, and low anterior hair line may be the only unique facial features [Jiang et al 2011].
Recurrent infections. Recurrent episodes of bacterial pneumonia are the most common infections in individuals with RMS [Iqbal et al 2022].
Integument. Cutaneous changes (hirsutism and acanthosis nigricans) typically appear later in childhood in individuals with RMS.
Cardiac manifestations. Hypertrophic cardiomyopathy is common and typically observed during the first decade of life. Neonatal hypertrophic cardiomyopathy was also reported in some individuals [Aftab et al 2022].
Renal manifestations. Enlarged kidneys are common. Hypercalcemia, nephrocalcinosis, and medullary sponge kidney are also reported as features of RMS [Chrzanowska et al 2023]. Nephrocalcinosis was reported in the vast majority of individuals [Simpkin et al 2014].
Liver/gastrointestinal manifestations. Hepatosplenomegaly occurs without liver or spleen dysfunction. Rectal prolapse was reported in some affected individuals [Joshi et al 2021].
Malignancy. Rare malignancies have been reported. Endometrial carcinoma was reported in a woman age 24 years treated with recombinant human IGF-1 (rhIGF-1) for severe insulin resistance [Jo et al 2013]. Ovarian granulosa cell tumor was reported in a girl age 35 months with severe insulin resistance treated with rhIGF-1 for 16 months [Weber et al 2014] and in a young girl who was untreated [Brisigotti et al 1993]. Because two of these individuals were treated with rhIGF-1, it is possible that the tumors resulted from an adverse effect of this treatment.
Prognosis. Complications of prolonged hyperglycemia and hyperinsulinemia are the major causes of death in individuals with RMS, usually during the second decade of life [Semple et al 2010]. However, survival can be into the third decade [Longo et al 2002, Musso et al 2004].
Genotype-Phenotype Correlations
There are no known genotype-phenotype correlations in INSR-SIRS.
Nomenclature
Leprechaunism is a synonym of Donohue syndrome.
Prevalence
Donohue syndrome is extremely rare, estimated at 1:1,000,000 [Desbois-Mouthon et al 1997].
To date, 96 individuals with Donohue syndrome and RMS have been molecularly diagnosed and reported [Ardon et al 2014, Ben Abdelaziz et al 2016, Termote et al 2016, Moore et al 2017, Chen et al 2018, Dagdeviren Cakir et al 2020, Al-Kandari et al 2021, Dos Santos et al 2021, Joshi et al 2021, Lai et al 2021, Zhou et al 2021, Novoa et al 2022, Chrzanowska et al 2023, Rojas Velazquez et al 2024]; about ten additional individuals have Donohue syndrome and RMS based on clinical diagnosis.
Genetically Related (Allelic) Disorders
Type A insulin resistance (OMIM 610549). Heterozygous or, less commonly, biallelic pathogenic variants in INSR are known to be associated with type A insulin resistance, especially when the pathogenic variant is in the intracellular domain [Takahashi et al 1997, Longo et al 2002, Jiang et al 2011, Zhou et al 2021]. Type A insulin resistance is characterized by hyperandrogenism (manifest as hirsutism, acne, and amenorrhea) and signs of insulin resistance (e.g., acanthosis nigricans and diabetes) without obesity, and most commonly comes to attention around the time of puberty due to findings that resemble polycystic ovarian syndrome. Of note, males can also be affected. The insulin resistance, which is milder than that of INSR-related severe insulin resistance syndrome, can be treated. Growth and cognitive abilities are normal [Musso et al 2004, Semple et al 2010].
Differential Diagnosis
The differential diagnosis of INSR-related severe insulin resistance syndrome (INSR-SIRS) includes many rare disorders with hirsutism, severe growth deficiency, and developmental delay with other syndromic features, some of which are summarized in Table 3.
Table 3.
Selected Disorders of Interest in the Differential Diagnosis of INSR-Related Severe Insulin Resistance Syndrome
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with INSR-related severe insulin resistance syndrome (INSR-SIRS), the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
INSR-Related Severe Insulin Resistance Syndrome: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
There is no cure for INSR-SIRS. Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 5).
Table 5.
INSR-Related Severe Insulin Resistance Syndrome: Treatment of Manifestations
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 6 are recommended [Kushchayeva et al 2019, Perge et al 2020, Huang-Doran et al 2021, Jansen et al 2022].
Table 6.
INSR-Related Severe Insulin Resistance Syndrome: Recommended Surveillance
Agents/Circumstances to Avoid
In individuals with Donohue syndrome, avoid the following:
- Agents that cause hypoglycemia
- Prolonged fasting
- Contact with persons with a contagious disease
In individuals with RMS, avoid the following:
- Agents that cause hyperglycemia
- High-carbohydrate diet
- Contact with persons with a contagious disease
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
There are no controlled trials in INSR-SIRS; thus, the two therapies discussed in this section are for individuals with INSR pathogenic variants based on clinical experience, case series, and expert opinion.
Recombinant Human Insulin-like Growth Factor 1 (rhIGF-1)
The rationale for using rhIGF-1 to treat severe insulin resistance syndromes is based on observations of its direct effects on carbohydrate metabolism. In humans, infusion of rhIGF-1 suppresses hepatic glucose production, stimulates peripheral glucose uptake in muscle, and – despite a significant reduction in circulating insulin levels – causes hypoglycemia. The insulin-like growth factor 1 receptor (IGF1R) and the insulin receptor (INSR) share 60% homology and very similar intracellular activity [Weber et al 2014].
Treatment regimen. Although many case reports of severe insulin resistance syndromes describe treatment with rhIGF-1, there is no standard protocol regarding this treatment. In fact, the treatment regimens either differed [McDonald et al 2007, de Kerdanet et al 2015] or were not reported.
Three treatment regimens for rhIGF-1 therapy in children with severe insulin resistance were reported: subcutaneous injections two to four times a day, continuous subcutaneous infusion via insulin pump, and intravenous infusion [Backeljauw et al 1994]. Anecdotal experience suggests that the use of continuous rhIGF-1 infusion is more beneficial than divided doses. Doses ranged from 80 to 1,120 µg/kg per day [McDonald et al 2007].
Outcome of treatment. In most treated children:
- The metabolic state improved and levels of glucose, insulin, and glycosylated hemoglobin decreased.
- Growth parameters improved [Nakae et al 1998].
- Renal tubular dysfunction improved [Hovnik et al 2013].
In some children treated with rhIGF-1, the degree of cardiomyopathy improved and survival was prolonged [McDonald et al 2007, de Kerdanet et al 2015, Carmody et al 2016], whereas in others no improvement was observed [Musso et al 2004, Grasso et al 2013].
Potential side effects include severe hypoglycemia and soft tissue overgrowth.
Given a lack of evidence, it is unclear whether the following two instances were side effects of the treatment or the hyperinsulinemia itself:
- Endometrial carcinoma in a woman age 24 years treated with rhIGF-1 for severe insulin resistance (called Donohue syndrome by the authors, but clinically more likely RMS) [Jo et al 2013]
- Granulosa cell tumor of the ovary in a girl age 35 months with severe insulin resistance treated with rhIGF-1 for 16 months [Weber et al 2014]
To summarize the experience with rhIGF-1 treatment in severe insulin resistance syndromes: (1) its benefit is not well established; and (2) it is more likely to be effective in individuals with less severe insulin resistance, as the few individuals with prolonged survival with rhIGF-1 treatment had milder phenotypes [Carmody et al 2016]. To date, rhIGF-1 appears to be the best treatment option; it is thus reasonable to consider in any individual with severe syndromic insulin resistance.
Metreleptin (Recombinant Human Leptin)
Metreleptin is approved by the FDA for treatment of congenital or acquired generalized lipodystrophy.
Leptin replacement normalized blood lipids (i.e., reduced triglycerides and increased high-density lipoproteins [HDL]) and reduced insulin and glucose levels in syndromes with leptin deficiency. Syndromes with leptin deficiency are characterized by insulin resistance, hyperglycemia, dyslipidemia, endocrine disruptions, and fatty liver disease [Paz-Filho et al 2015] and include lipodystrophy syndromes, hypothalamic amenorrhea, anorexia nervosa, and congenital leptin deficiency.
After one year of treatment with metreleptin (along with other medications including insulin, metformin, and pioglitazone), individuals with RMS showed improvement in all of the following: serum glucose levels, glycosylated hemoglobin (HbA1c) levels, insulinemia, insulin dose required, caloric intake, and body fat mass [Cochran et al 2004, Brown et al 2013].
Metreleptin alters the natural history of rising HbA1c in RMS, leading to lower HbA1c throughout long-term follow up. Improved glycemia with metreleptin is likely attributable to appetite suppression and lower body mass index (BMI). Lower BMI after metreleptin may also worsen growth hormone resistance in RMS, resulting in a null effect on IGF-1 and growth despite improved glycemia [Okawa et al 2022].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
INSR-related severe insulin resistance syndrome (INSR-SIRS) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are presumed to be heterozygous for an INSR pathogenic variant.
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for an INSR pathogenic variant and to allow reliable recurrence risk assessment.
- If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Maassen et al 2003, Kawashima et al 2013]. If the proband appears to have homozygous pathogenic variants (i.e., the same two pathogenic variants), additional possibilities to consider include:
- A single- or multiexon deletion in the proband that was not detected by sequence analysis and that resulted in the artifactual appearance of homozygosity;
- Uniparental isodisomy for the parental chromosome with the pathogenic variant that resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes are usually asymptomatic but may have features of the allelic disorder type A insulin resistance (see Genetically Related Disorders). Females who are heterozygous for an INSR pathogenic variant are at increased risk for gestational diabetes (see Family planning).
Sibs of a proband
- If both parents are known to be heterozygous for an INSR pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting biallelic pathogenic variants and being affected with INSR-SIRS, a 50% chance of inheriting one pathogenic variant and being heterozygous, and a 25% chance of inheriting neither of the familial INSR pathogenic variants.
- Heterozygotes are usually asymptomatic but may have features of the allelic disorder type A insulin resistance (see Genetically Related Disorders). Females who are heterozygotes for an INSR pathogenic variant are at increased risk for gestational diabetes (see Family planning).
Offspring of a proband. Individuals with INSR-SIRS are not known to reproduce.
Other family members. Each sib of the proband's parents is at a 50% risk of being heterozygous for an INSR pathogenic variant.
Heterozygote Detection
Heterozygote testing for at-risk relatives requires prior identification of the INSR pathogenic variants in the family.
Heterozygotes are usually asymptomatic but may have features of the allelic disorder type A insulin resistance. Females who are heterozygous for an INSR pathogenic variant are at increased risk for gestational diabetes (see Family planning).
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are heterozygous or are at risk of being heterozygous.
- Heterozygote testing should be considered for the reproductive partners of known heterozygotes, particularly if both partners are of the same ancestry. The prevalence of heterozygotes for the pathogenic variant c.167T>C is high among Druze in Israel [Falik Zaccai et al 2014].
- Females who are heterozygous for an INSR pathogenic variant are at increased risk for gestational diabetes and require monitoring for glucose intolerance before and during pregnancy [Kleijer et al 2006]. See ElSayed et al [2023] for management recommendations.
Prenatal Testing and Preimplantation Genetic Testing
Once the INSR pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- National Organization for Rare Disorders (NORD)
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
INSR-Related Severe Insulin Resistance Syndrome: Genes and Databases
Table B.
OMIM Entries for INSR-Related Severe Insulin Resistance Syndrome (View All in OMIM)
Molecular Pathogenesis
INSR encodes the insulin receptor, a member of the superfamily of transmembrane receptor tyrosine kinases. The INSR preprotein, consisting of 1,382 amino acids, is proteolytically processed to generate the alpha and beta subunits of INSR, a heterotetrameric glycoprotein. The 731-amino acid alpha subunit, which is external to the plasma membrane, contains the insulin-binding region. The alpha subunit is linked by disulfide bonds to the 620-amino acid beta subunit, which includes a 194-amino acid extracellular domain, a 23-amino acid membrane-spanning segment, and a 403-amino acid cytoplasmic segment that has intrinsic tyrosine kinase activity [Seino et al 1989].
When insulin binds to the alpha subunit, the beta subunit undergoes autophosphorylation, which activates the insulin-signaling pathway regulating glucose uptake and release as well as the synthesis and storage of carbohydrates, lipids, and protein.
Taylor et al [1991] described five classes of INSR pathogenic variants that:
- Impair synthesis of the receptors;
- Impair transport of receptors to the cell membrane;
- Decrease receptor affinity for insulin;
- Reduce the tyrosine kinase activity of the receptor intracellular domain;
- Accelerate receptor degradation [Porter & Barrett 2005].
In severe insulin resistance, reduced intracellular insulin signaling causes hyperglycemia. The hypoglycemia in INSR-related severe insulin resistance syndrome is thought to be a consequence of late action of insulin-like growth factor 1 (IGF-1) [Kawashima et al 2013]. Insulin resistance causes alterations in the expression of genes encoding growth factors and apoptosis [Iovino et al 2014].
Mechanism of disease causation. Loss of function
Table 7.
Notable INSR Pathogenic Variants
Chapter Notes
Author Notes
Author research interests:
- Searching for genes responsible for various rare genetic disorders and investigating the clinical, biochemical, and molecular basis for each disorder by studying the related protein function and biologic pathway. Diseases currently of particular interest include neurogenetic diseases, aplasia of distal phalanges with juvenile breast hypertrophy (MDN), osteogenesis imperfecta, and hereditary spastic paraparesis and cardiomyopathies.
- Identification of new pathogenic variants, genes, and proteins involved in NER-type DNA repair mechanisms and understanding their cellular function and their role in premature aging and cancer. In addition, the establishment of new diagnostic procedures for the screening of causative pathogenic variants in affected individuals in Israel and the Middle East.
- Development of methods of genetic counseling tailored to kindreds at high risk for genetic disorders, in an attempt to raise awareness and to prevent and minimize the births of affected individuals. Also, early identification of affected newborns is critical for provision of prompt and effective treatment.
- Genetics of pain. Our interest in this field is manifested in the study of women with vulvodynia (pain during sexual intercourse). This phenomenon is evident within families, and genetic associations have been found. With the collaboration of Professor J Bornstein, we are studying possible genetic associations that relate to biochemical pathways involved in pain regulation among a large cohort of women affected with vulvodynia.
Dr Falik Zaccai's web page
Author History
Shani Ben Harouch, MD; Bar-Ilan University (2018-2024)
Tzipora C Falik Zaccai, MD (2018-present)
Aharon Klar, MD (2018-present)
Aviv Mesika (2024-present)
Revision History
- 25 April 2024 (sw) Comprehensive updated posted live
- 25 January 2018 (bp) Review posted live
- 14 March 2016 (sbh) Original submission
References
Literature Cited
- Aftab S, Shaheen T, Asif R, Anjum MN, Saeed A, Manzoor J, Cheema HA. Management challenges of Rabson Mendenhall syndrome in a resource limited country: a case report. J Pediatr Endocrinol Metab. 2022;35:1429-32. [PubMed: 36106528]
- Al-Kandari H, Al-Abdulrazzaq D, Al-Jaser F, Al-Mulla F, Davidsson L. Rabson-Mendenhall Syndrome in a brother-sister pair in Kuwait: Diagnosis and 5 year follow up. Prim Care Diabetes. 2021;15:175-7. [PubMed: 32843252]
- Angelidi AM, Filippaios A, Mantzoros CS. Severe insulin resistance syndromes. J Clin Invest. 2021;131:e142245. [PMC free article: PMC7880309] [PubMed: 33586681]
- Ardon O, Procter M, Tvrdik T, Longo N, Mao R. Sequencing analysis of insulin receptor defects and detection of two novel mutations in INSR gene. Mol Genet Metab Rep. 2014;1:71-84. [PMC free article: PMC5121292] [PubMed: 27896077]
- Backeljauw PF, Alves C, Eidson M. Effect of intravenous insulin-like growth factor I in two patients with leprechaunism. Pediatr Res. 1994;36:749-54. [PubMed: 7534902]
- Bamborschke D, Özdemir Ö, Kreutzer M, Motameny S, Thiele H, Kribs A, Dötsch J, Altmüller J, Nürnberg P, Cirak S. Ultra-rapid emergency genomic diagnosis of Donahue syndrome in a preterm infant within 17 hours. Am J Med Genet A. 2021;185:90-6. [PubMed: 33048476]
- Baqir ZS, Al-Lawati TT, Al Hussaini SO, Al-Sinani A, Al-Said K, Al-Rashdi I. A novel leprechaunism mutation, Cys807Arg, in an Arab infant: a rare cause of hypoglycaemia. Paediatr Int Child Health. 2012;32:183-5. [PubMed: 22824672]
- Bathi RJ, Parveen S, Mutalik S. Rabson-Mendenhall syndrome : two case reports and a brief review of the literature. Odontology. 2010;98:89-96. [PubMed: 20155514]
- Ben Abdelaziz R, Ben Chehida A, Azzouz H, Boudabbous H, Lascols O, Ben Turkia H, Tebib N. A novel homozygous missense mutation in the insulin receptor gene results in an atypical presentation of Rabson-Mendenhall syndrome. Eur J Med Genet. 2016;59:16-9. [PubMed: 26691667]
- Brisigotti M, Fabbretti G, Pesce F, Gatti R, Cohen A, Parenti G, Callea F. Congenital bilateral juvenile granulosa cell tumor of the ovary in leprechaunism: a case report. Pediatr Pathol. 1993;13:549-58. [PubMed: 8247952]
- Brown RJ, Cochran E, Gorden P. Metreleptin improves blood glucose in patients with insulin receptor mutations. J Clin Endocrinol Metab. 2013;98:E1749-56. [PMC free article: PMC3816267] [PubMed: 23969187]
- Brown RJ, Joseph J, Cochran E, Gewert C, Semple R, Gorden P. Type B insulin resistance masquerading as ovarian hyperthecosis. J Clin Endocrinol Metab. 2017;102:1789-91. [PMC free article: PMC5470776] [PubMed: 27911591]
- Carmody D, Ladsaria SS, Buikema RK, Semple RK, Greeley SAW. Successful rhIGF1 treatment for over 5 years in a patient with severe insulin resistance due to homozygous insulin receptor mutation. Diabet Med. 2016;33:e8-e12. [PMC free article: PMC4751063] [PubMed: 26262567]
- Carrasco de la Fuente M, Barrio Castellanos R, Alonso Blanco M, de la Calle Blasco H. Long survival in Rabson-Mendenhall syndrome. Diabetes Res Clin Pract. 2010;89:e17-18. [PubMed: 20627358]
- Chen X, Wang H, Wu B, Dong X, Liu B, Chen H, Lu Y, Zhou W, Yang L. One Novel 2.43Kb deletion and one single nucleotide mutation of the INSR gene in a Chinese neonate with Rabson-Mendenhall syndrome. J Clin Res Pediatr Endocrinol. 2018;10:183-7. [PMC free article: PMC5985390] [PubMed: 29082893]
- Chong YH, Taylor BJ, Wheeler BJ. Renal manifestations of severe Rabson-Mendenhall syndrome: a case report. J Diabetes Metab Disord. 2013;12:7. [PMC free article: PMC3598197] [PubMed: 23497647]
- Chrzanowska J, Skarul J, Zubkiewicz-Kucharska A, Borowiec M, Zmyslowska A. Incomplete phenotypic presentation in a girl with rare Rabson-Mendenhall syndrome. Acta Diabetol. 2023;60:449-53. [PMC free article: PMC9931845] [PubMed: 36331627]
- Cochran E, Young JR, Sebring N, DePaoli A, Oral EA, Gorden P. Efficacy of recombinant methionyl human leptin therapy for the extreme insulin resistance of the Rabson-Mendenhall syndrome. J Clin Endocrinol Metab. 2004;89:1548-54. [PubMed: 15070911]
- Dagdeviren Cakir A, Saidov S, Turan H, Ceylaner S, Özer Y, Kutlu T, Ercan O, Evliyaoglu O. Two novel variants and one previously reported variant in the insulin receptor gene in two cases with severe insulin resistance syndrome. Mol Syndromol. 2020;11:90-6. [PMC free article: PMC7325121] [PubMed: 32655340]
- de Bock M, Hayes I, Semple R. Donohue syndrome. J Clin Endocrinol Metab. 2012;97:1416-17. [PubMed: 22399506]
- de Kerdanet M, Caron-Debarle M, Nivot S, Gaillot T, Lascols O, Fremont B, Bonnaure-Mallet M, Gie S, Massart C, Capeau J. Ten-year improvement of insulin resistance and growth with recombinant human insulin-like growth factor 1 in a patient with insulin receptor mutations resulting in leprechaunism. Diabetes Metab. 2015;41:331-7. [PubMed: 25465274]
- Desbois-Mouthon C, Girodon E, Ghanem N, Caron M, Pennerath A, Conteville P, Magre J, Besmond C, Goossens M, Capeau J, Amselem S. Molecular analysis of the insulin receptor gene for prenatal diagnosis of leprechaunism in two families. Prenat Diagn. 1997;17:657-63. [PubMed: 9249867]
- Dos Santos SS, Ramaldes LA, Gabbay MAL, Moises RCS, Dib SA. Use of a sodium-glucose cotransporter 2 inhibitor, empagliflozin, in a patient with Rabson-Mendenhall syndrome. Horm Res Paediatr. 2021;94:313-6. [PubMed: 34551418]
- Elders MJ, Schedewie HK, Olefsky J, Givens B, Char F, Bier DM, Baldwin D, Fiser RH, Seyedabadi S, Rubenstein A. Endocrine-metabolic relationships in patients with leprechaunism. J Natl Med Assoc. 1982;74:1195-210. [PMC free article: PMC2561419] [PubMed: 7154104]
- ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, Collins BS, Hilliard ME, Isaacs D, Johnson EL, Kahan S, Khunti K, Leon J, Lyons SK, Perry ML, Prahalad P, Pratley RE, Jeffrie Seley J, Stanton RC, Gabbay RA, on behalf of the American Diabetes Association. 15. Management of diabetes in pregnancy: standards of care in diabetes-2023. Diabetes Care. 2023;46(Suppl 1):S254-66. [PMC free article: PMC9810465] [PubMed: 36507645]
- Falik Zaccai TC, Kalfon L, Klar A, Elisha M Ben, Hurvitz H, Weingarten G, Chechik E, Fleisher Sheffer V, Haj Yahya R, Meidan G, Gross-Kieselstein E, Bauman D, Hershkovitz S, Yaron Y, Orr-Urtreger A, Wertheimer E. Two novel mutations identified in familial cases with Donohue syndrome. Mol Genet Genomic Med. 2014;2:64-72. [PMC free article: PMC3907912] [PubMed: 24498630]
- Geffner ME, Kaplan SA, Bersch N, Lippe BM, Smith WG, Nagel RA, Santulli TV Jr, Li CH, Golde DW. Leprechaunism: in vitro insulin action despite genetic insulin resistance. Pediatr Res. 1987;22:286-91. [PubMed: 3309859]
- Gosavi S, Sangamesh S, Ananda Rao A, Patel S, Hodigere VC. Insulin, insulin everywhere: a rare case report of Rabson-Mendenhall syndrome. Cureus. 2021;13:e13126. [PMC free article: PMC7936575] [PubMed: 33728143]
- Grasso V, Colombo C, Favalli V, Galderisi A, Rabbone I, Gombos S, Bonora E, Massa O, Meschi F, Cerutti F, Iafusco D, Bonfanti R, Monciotti C, Barbetti F. Six cases with severe insulin resistance (SIR) associated with mutations of insulin receptor: Is a Bartter-like syndrome a feature of congenital SIR? Acta Diabetol. 2013;50:951-7. [PubMed: 23824322]
- Hovnik T, Bratanič N, Podkrajšek KT, Kovač J, Paro D, Podnar T, Bratina N, Battelino T. Severe progressive obstructive cardiomyopathy and renal tubular dysfunction in Donohue syndrome with decreased insulin receptor autophosphorylation due to a novel INSR mutation. Eur J Pediatr. 2013;172:1125-9. [PubMed: 23229189]
- Huang-Doran I, Kinzer AB, Jimenez-Linan M, Thackray K, Harris J, Adams CL, de Kerdanet M, Stears A, O'Rahilly S, Savage DB, Gorden P, Brown RJ, Semple RK. Ovarian hyperandrogenism and response to gonadotropin-releasing hormone analogues in primary severe insulin resistance. J Clin Endocrinol Metab. 2021;106:2367-83. [PMC free article: PMC8277216] [PubMed: 33901270]
- Iovino S, Burkart AM, Kriauciunas K, Warren L, Hughes KJ, Molla M, Lee YK, Patti ME, Kahn CR. Genetic insulin resistance is a potent regulator of gene expression and proliferation in human iPS cells. Diabetes. 2014;63:4130-42. [PMC free article: PMC4238001] [PubMed: 25059784]
- Iqbal J, Jiang HL, Wu HX, Li L, Zhou YH, Hu N, Xiao F, Wang T, Xu SN, Zhou HD. Hereditary severe insulin resistance syndrome: Pathogenesis, pathophysiology, and clinical management. Genes Dis. 2022;10:1846-56. [PMC free article: PMC10363564] [PubMed: 37492723]
- Jansen M, Algül S, Bosman LP, Michels M, van der Velden J, de Boer RA, van Tintelen JP, Asselbergs FW, Baas AF. Blood-based biomarkers for the prediction of hypertrophic cardiomyopathy prognosis: a systematic review and meta-analysis. ESC Heart Fail. 2022;9:3418-34. [PMC free article: PMC9715795] [PubMed: 35842920]
- Jiang S, Fang Q, Zhang F, Wan H, Zhang R, Wang C, Bao Y, Zhang L, Ma X, Lu J, Gao F, Xiang K, Jia W. Functional characterization of insulin receptor gene mutations contributing to Rabson-Mendenhall syndrome - phenotypic heterogeneity of insulin receptor gene mutations. Endocr J. 2011;58:931-40. [PubMed: 21869538]
- Jo W, Sudo S, Nakamura A, Endo D, Konno Y, Ishizu K, Tajima T. Development of endometrial carcinoma in a patient with leprechaunism (Donohue syndrome). Clin Pediatr Endocrinol. 2013;22:33-8. [PMC free article: PMC3756185] [PubMed: 23990696]
- Joshi SR, Pendyala GS, Shah P, Pustake B, Mopagar V, Padmawar N. Severe insulin resistance syndrome - A rare case report and review of literature. Natl J Maxillofac Surg. 2021;12:100-5. [PMC free article: PMC8191564] [PubMed: 34188410]
- Kawashima Y, Nishimura R, Utsunomiya A, Kagawa R, Funata H, Fujimoto M, Hanaki K, Kanzaki S. Leprechaunism (Donohue syndrome): a case bearing novel compound heterozygous mutations in the insulin receptor gene. Endocr J. 2013;60:107-12. [PubMed: 22972224]
- Kim D, Cho SY, Sohn YB, Kwon M. Two novel insulin receptor gene mutations in a patient with Rabson-Mendenhall syndrome: the first Korean case confirmed by biochemical and molecular evidence. J Korean Med Sci. 2012;27:565-8. [PMC free article: PMC3342552] [PubMed: 22563226]
- Klammt J, Kiess W, Pfäffle R. IGF1R mutations as cause of SGA. Best Pract Res Clin Endocrinol Metab. 2011;25:191-206. [PubMed: 21396585]
- Kleijer WJ, van der Sterre MLT, Garritsen VH, Raams A, Jaspers NGJ. Prenatal diagnosis of the Cockayne syndrome: survey of 15 years experience. Prenat Diagn. 2006;26:980-4. [PubMed: 16941719]
- Kushchayeva YS, Kushchayev SV, Startzell M, Cochran E, Auh S, Dai Y, Lightbourne M, Skarulis M, Brown RJ. Thyroid abnormalities in patients with extreme insulin resistance syndromes. J Clin Endocrinol Metab. 2019;104:2216-28. [PMC free article: PMC6482021] [PubMed: 30657911]
- Lai L, Mikhchi A, Ryabets-Lienhard A, Geffner ME, Cheung C, Guiffre D. Reversible severe ovarian enlargement in an infant with significant insulin resistance. Radiol Case Rep. 2021;16:1760-5. [PMC free article: PMC8111256] [PubMed: 34007398]
- Longo N, Wang Y, Smith SA, Langley SD, DiMeglio LA, Giannella-Neto D. Genotype-phenotype correlation in inherited severe insulin resistance. Hum Mol Genet. 2002;11:1465-75. [PubMed: 12023989]
- Maassen JA, Tobias ES, Kayserilli H, Tukel T, Yuksel-Apak M, D'Haens E, Kleijer WJ, Féry F, van der Zon GC. Identification and functional assessment of novel and known insulin receptor mutations in five patients with syndromes of severe insulin resistance. J Clin Endocrinol Metab. 2003;88:4251-7. [PubMed: 12970295]
- McDonald A, Williams RM, Regan FM, Semple RK, Dunger DB. IGF-I treatment of insulin resistance. Eur J Endocrinol 2007;157:S51-6. [PubMed: 17785698]
- Metwalley KA, Farghaly HS, Maxi LM. Donohue syndrome in an Egyptian infant: a case report. Case Rep Perinat Med. 2022;12.
- Moore MM, Bailey AM, Flannery AH, Baum RA. Treatment of diabetic ketoacidosis with intravenous U-500 insulin in a patient with Rabson-Mendenhall syndrome: a case report. J Pharm Pract. 2017;30:468-75. [PubMed: 27112737]
- Moreira RO, Zagury RL, Nascimento TS. Multidrug therapy in a patient with Rabson-Mendenhall syndrome. Diabetologia. 2010;53:2454-5. [PubMed: 20711714]
- Musso C, Cochran E, Moran SA, Skarulis MC, Oral EA, Taylor S, Gorden P. Clinical course of genetic diseases of the insulin receptor (type A and Rabson-Mendenhall syndromes): a 30-year prospective. Medicine (Baltimore). 2004;83:209-22. [PubMed: 15232309]
- Nakae J, Kato M, Murashita M, Shinohara N, Tajima T, Fujieda K. Long-term effect of recombinant human insulin-like growth factor I on metabolic and growth control in a patient with leprechaunism. J Clin Endocrinol Metab. 1998;83:542-9. [PubMed: 9467572]
- Nicolescu CR, Cremillieux C, Stephan JL. Duodenogastric intussusception in a 14-week-old infant with Donohue syndrome: case study. Case Rep Pediatr. 2023;2023:7799234. [PMC free article: PMC10599843] [PubMed: 37885901]
- Novoa MP, Hernandez JF, Florez J. Rabson-Mendenhall syndrome in a 6-year-old girl. Eur J Pediat Dermatol. 2022;32:245-7.
- Okawa MC, Cochran E, Lightbourne M, Brown RJ. Long-term effects of metreleptin in Rabson-Mendenhall syndrome on glycemia, growth, and kidney function. J Clin Endocrinol Metab. 2022;107:e1032-e1046. [PMC free article: PMC8852213] [PubMed: 34718628]
- Parker VE, Semple RK. Genetics in endocrinology: genetic forms of severe insulin resistance: what endocrinologists should know. Eur J Endocrinol. 2013;169:R71-80. [PMC free article: PMC4359904] [PubMed: 23857978]
- Paz-Filho G, Mastronardi CA, Licinio J. Leptin treatment: facts and expectations. Metabolism. 2015;64:146-56. [PubMed: 25156686]
- Perge K, Massoud M, Gauthier-Moulinier H, Lascols O, Pangaud N, Villanueva C, Pons L. Intrauterine growth restriction and hypertrophic cardiomyopathy as prenatal ultrasound findings in a case of leprechaunism. Mol Syndromol. 2020;11:223-7. [PMC free article: PMC7675228] [PubMed: 33224016]
- Porter JR, Barrett TG. Monogenic syndromes of abnormal glucose homeostasis: clinical review and relevance to the understanding of the pathology of insulin resistance and B cell failure. J Med Genet. 2005;42:893-902. [PMC free article: PMC1735963] [PubMed: 15772126]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rojas Velazquez MN, Blanco F, Ayala-Lugo A, Franco L, Jolly V, Di Tore D, Martínez de Lapiscina I, Janner M, Flück CE, Pandey AV. A novel mutation in the INSR gene causes severe insulin resistance and Rabson-Mendenhall syndrome in a Paraguayan patient. Int J Mol Sci. 2024;25:3143. [PMC free article: PMC10970221] [PubMed: 38542117]
- Rojek A, Wikiera B, Noczynska A, Niedziela M. Syndrome of congenital insulin resistance caused by a novel INSR gene mutation. J Clin Res Pediatr Endocrinol. 2023;15:312-7 [PMC free article: PMC10448552] [PubMed: 34965699]
- Seino S, Seinot M, Nishi S, Bell GI. Structure of the human insulin receptor gene and characterization of its promoter. Proc Natl Acad Sci U S A. 1989;86:114-8. [PMC free article: PMC286414] [PubMed: 2911561]
- Semple RK, Cochran EK, Soos MA, Burling KA, Savage DB, Gorden P, O'Rahilly S. Plasma adiponectin as a marker of insulin receptor dysfunction: clinical utility in severe insulin resistance. Diabetes Care. 2008;31:977-9. [PubMed: 18299442]
- Semple RK, Savage DB, Cochran EK, Gorden P, O'Rahilly S. Genetic syndromes of severe insulin resistance. Endocr Rev. 2011;32:498-514. [PubMed: 21536711]
- Semple RK, Sleigh A, Murgatroyd PR, Adams CA, Bluck L, Jackson S, Vottero A, Kanabar D, Charlton-Menys V, Durrington P, Soos MA, Carpenter TA, Lomas DJ, Cochran EK, Gorden P, O'Rahilly S, Savage DB. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest. 2009;119:315-22 [PMC free article: PMC2631303] [PubMed: 19164855]
- Semple RK, Williams RM, Dunger DB. What is the best management strategy for patients with severe insulin resistance? Clin Endocrinol (Oxf). 2010;73:286-90. [PubMed: 20455892]
- Siala-Sahnoun O, Dhieb D, Ben Thabet A, Hmida N, Belguith N, Fakhfakh F. First molecular diagnosis of Donohue syndrome in Africa: novel unusual insertion/deletion mutation in the INSR gene. Mol Biol Rep. 2016;43:165-73. [PubMed: 26874853]
- Simpkin A, Cochran E, Cameron F, Dattani M, de Bock M, Dunger DB, Forsander G, Guran T, Harris J, Isaac I, Hussain K, Kleta R, Peters C, Tasic V, Williams R, Yap Kok Peng F, O'Rahilly S, Gorden P, Semple RK, Bockenhauer D. Insulin receptor and the kidney: nephrocalcinosis in patients with recessive INSR mutations. Nephron Physiol. 2014;128:55-61. [PMC free article: PMC4369119] [PubMed: 25358339]
- Sinnarajah K, Dayasiri MB, Dissanayake ND, Kudagammana ST, Jayaweera AH. Rabson Mendenhall syndrome caused by a novel missense mutation. Int J Pediatr Endocrinol. 2016;2016:21. [PMC free article: PMC5114824] [PubMed: 27891155]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Takahashi Y, Kadowaki H, Momomura K, Fukushima Y, Orban T, Okai T, Taketani Y, Akanuma Y, Yazaki Y, Kadowaki T. A homozygous kinase-defective mutation in the insulin receptor gene in a patient with leprechaunism. Diabetologia. 1997;40:412-20. [PubMed: 9112018]
- Taylor SI, Accili D, Cama A, Kadowaki H, Kadowaki T, Imano E, Sierra ML. Mutations in the insulin receptor gene in patients with genetic syndromes of insulin resistance. Adv Exp Med Biol. 1991;293:197-213. [PubMed: 1767731]
- Termote JU, Breur JM, de Vroede MA. Hypertrophic cardiomyopathy in Donohue syndrome. Cardiol Young. 2016;26:815-8. [PubMed: 26555333]
- Tuthill A, Semple RK, Day R, Soos MA, Sweeney E, Seymour PJ, Didi M, O'Rahilly S. Functional characterization of a novel insulin receptor mutation contributing to Rabson-Mendenhall syndrome. Clin Endocrinol (Oxf). 2007;66:21-6. [PubMed: 17201797]
- Vakili K, Rezaei N. Rabson-Mendenhall syndrome. In: Rezaei N (ed), Genetic Syndromes. Springer, Cham; 2023.
- Weber DR, Stanescu DE, Semple R, Holland C, Magge SN. Continuous subcutaneous IGF-1 therapy via insulin pump in a patient with Donohue syndrome. J Pediatr Endocrinol Metab. 2014;27:1237-41. [PMC free article: PMC4535795] [PubMed: 25153212]
- Wei C, Burren CP. Diagnostic and management challenges from childhood, puberty through to transition in severe insulin resistance due to insulin receptor mutations. Pediatr Diabetes. 2017;18:835-8. [PubMed: 28093873]
- Zhou Q, Yu J, Yuan X, Wang C, Zhu Z, Zhang A, Gu W. Clinical and functional characterization of novel INSR variants in two families with severe insulin resistance syndrome. Front Endocrinol (Lausanne). 2021;12:606964. [PMC free article: PMC8117416] [PubMed: 33995269]
Publication Details
Author Information and Affiliations
Shaare Zedek Medical Center
Hadassah Medical School
Hebrew University
Jerusalem, Israel
Publication History
Initial Posting: January 25, 2018; Last Update: April 25, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Mesika A, Klar A, Falik Zaccai TC. INSR-Related Severe Insulin Resistance Syndrome. 2018 Jan 25 [Updated 2024 Apr 25]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.