Clinical Description
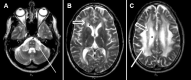
LMNB1-related autosomal dominant leukodystrophy (ADLD) is a slowly progressive neurologic disorder of central nervous system white matter. Adults show autonomic dysfunction, the first evidence of the disorder, in the fourth to fifth decade of life, followed by pyramidal and cerebellar abnormalities resulting in spasticity, ataxia, and tremor [Finnsson et al 2015].
To date, at least 33 families have been identified with a pathogenic variant in LMNB1 [Eldridge et al 1984, Quattrocolo et al 1997, Coffeen et al 2000, Marklund et al 2006, Padiath et al 2006, Meijer et al 2008, Brussino et al 2009, Schuster et al 2011, Dos Santos et al 2012, Molloy et al 2012, Potic et al 2013, Dai et al 2017, Sandoval-Rodríguez et al 2017, Mezaki et al 2018, Zhang et al 2019]. The following description of the phenotypic features associated with this condition is based on these reports.
Autonomic dysfunction, a nearly universal feature [Padiath & Fu 2010], includes bladder dysfunction, constipation, postural hypotension, erectile dysfunction, and (less often) impaired sweating. Symptoms of autonomic dysfunction may be the initial disease presentation and can precede the motor and cerebellar manifestations by months to years.
Pyramidal signs and symptoms usually develop after manifestations of autonomic dysfunction; however, gait difficulties have been reported as the first manifestations [Finnsson et al 2015]. Pyramidal manifestations include signs of upper motor neuron dysfunction, often more prominent in the lower extremities (e.g., spastic weakness, hypertonia, clonus, brisk deep tendon reflexes, and bilateral Babinski signs). Spasticity can cause muscle pain and joint contractures. Over time, pyramidal dysfunction extends to the upper extremities.
Cerebellar signs that typically appear at the same time as the pyramidal signs include gait ataxia, dysdiadochokinesia, intention tremor, dysmetria, and nystagmus. Upper extremity postural tremor accompanied by neck tremor (resulting in head titubation) or jaw tremor (affecting speech and chewing) can also be seen [Schwankhaus et al 1988].
Cognitive function is usually preserved or mildly impaired early in the disease course; however, dementia and psychiatric manifestations can occur as late manifestations [Dos Santos et al 2012, Finnsson et al 2015].
Additional features
Neurophysiologic studies are normal (electromyogram, visual evoked potentials, nerve conduction studies) or demonstrate nonspecific findings (brain stem auditory evoked potentials, somatosensory evoked potentials) or both (electroencephalograms).
Laboratory findings (CSF analysis, measurements of catecholamines, peripheral nerve biopsy) are either normal or do not yield findings specific to this disorder.
Prognosis. Affected individuals may survive for decades after onset of symptoms. Motor manifestations are usually slowly progressive without acute exacerbations; however, some affected individuals report reversible worsening of cognitive function and gait with fever [Finnsson et al 2015].