Clinical Description
The clinical picture of LBSL consists of slowly progressive cerebellar ataxia, spasticity, and dorsal column dysfunction, involving the legs more than the arms. Most affected individuals have decreased position and vibration sense of the legs more than the arms, leading to increased difficulty walking in the dark. Manual dexterity becomes impaired to a variable degree.
To date, more than 100 individuals have been identified with biallelic pathogenic variants in DARS2. Large series [Steenweg et al 2011, van Berge et al 2014, Stellingwerff et al 2021] as well as multiple case reports [Köhler et al 2015, Shimojima et al 2017, Yahia et al 2018, Yelam et al 2019, N'Gbo N'Gbo Ikazabo et al 2020, Yazici Gencdal et al 2020] have been described. Prospective natural history data are currently not available. The following description of the phenotypic features associated with this condition is based on the cited reports.
Table 2.
Leukoencephalopathy with Brain Stem and Spinal Cord Involvement and Lactate Elevation: Frequency of Select Features
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
|
Normal early development
| 80% | Infantile form was recently identified [Stellingwerff et al 2021]; relative incidence is not clear yet but probably low. |
|
Loss of motor skills
| 100% | Core feature of LBSL |
|
Cerebellar ataxia
| Very common |
|
Spasticity
| Very common |
|
Axonal neuropathy
| Uncommon | ↓ or absent tendon reflexes, distal weakness, sensory loss |
|
Learning disability
| 20% | Most often learning disability; severe intellectual impairment is rare. |
|
Cognitive decline
| 20% | |
|
Epilepsy
| Uncommon | Usually easily controlled w/ASM |
ASM = anti-seizure medication
Variable severity. The range of disease spectrum includes:
Childhood onset. Most common form with slow progression. Most individuals with childhood onset become partially or fully wheelchair dependent in their teens, twenties, or later [
van Berge et al 2014].
Motor involvement. In most affected individuals, initial motor development is normal. Deterioration of motor skills usually starts in childhood or adolescence [Uluc et al 2008, van Berge et al 2014] and occasionally in infancy [Steenweg et al 2012] or adulthood [van Berge et al 2014]. Slowly progressive cerebellar ataxia and spasticity occur in most individuals. Axonal peripheral neuropathy may add to the distal weakness. The motor dysfunction involves the legs more than the arms.
In the individuals with antenatal or early-infantile onset, motor involvement is severe and typically no or very few motor milestones are achieved [Stellingwerff et al 2021].
Sensory involvement. There is severe dorsal column dysfunction with decreased position and vibration sense on neurologic examination, causing sensory ataxia especially in the legs. Evidence of an axonal neuropathy is found in some (not all) affected individuals, leading to decreased or absent tendon reflexes and distal sensory loss affecting the legs more than the arms [van der Knaap et al 2003, Távora et al 2007, Uluc et al 2008, Isohanni et al 2010]. However, true peripheral neuropathy is probably not common as neurophysiologic examination is often normal [van der Knaap et al 2003].
Speech. Dysarthria develops over time. Swallowing is generally not affected in individuals with childhood or adult onset [van Berge et al 2014].
Cognitive skills. Some have learning problems from early on, but most have normal intellectual capacity. Cognitive decline may occur and is usually slow [van der Knaap et al 2003, Serkov et al 2004]. Individuals with antenatal or early-infantile presentation typically have severe cognitive impairment [Stellingwerff et al 2021].
Epilepsy. Some affected individuals develop epilepsy. Seizures are infrequent and easily controlled with medication [van der Knaap et al 2003]. In individuals with antenatal and early-infantile onset, epilepsy is almost invariably present and often severe [Stellingwerff et al 2021].
Microcephaly. In individuals with antenatal or early-infantile onset, microcephaly is almost invariably present and often severe [Stellingwerff et al 2021]. See also .
Response to minor head trauma. Some affected individuals experience lowered consciousness, neurologic deterioration, and fever following minor head trauma [Serkov et al 2004]. Recovery is only partial.
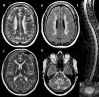
Routine laboratory tests, including cerebrospinal fluid (CSF) analysis, are usually normal. In a few individuals mild and inconsistent mild elevation of lactate concentration has been noted in blood or CSF or both. No published information is available.
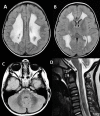
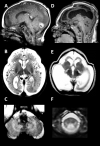
Neuropathologic findings have been described in two sibs with a severe variant of LBSL [Yamashita et al 2013]. Electron microscopy revealed vacuolar changes and myelin splitting in the affected white matter [Yamashita et al 2013]. Quantitative MR parameters are in line with these findings, with T2 hyperintense white matter showing increased fractional anisotropy and reduced apparent diffusion coefficient [Steenweg et al 2011].