Clinical Description
NOTCH3-related lateral meningocele syndrome (LMS) is characterized by multiple lateral spinal meningoceles, distinctive facial features, joint hyperextensibility, hypotonia, and skeletal, cardiac, and urogenital anomalies.
To date, 11 individuals have been identified with a pathogenic variant in NOTCH3 [Gripp et al 2015, Ejaz et al 2016, Brown et al 2017, Cappuccio et al 2020, Han et al 2022, Yamada et al 2022]. The following description of the phenotypic features associated with this condition is based on these reports.
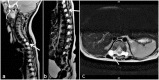
Multiple lateral spinal meningoceles (protrusions of the arachnoid and dura through the spinal foramina) are found in all affected individuals. Neurologic sequelæ of the meningoceles can include neurogenic bladder, paresthesia, back pain, and/or paraparesis depending on size and location. Other neurologic findings can include Chiari I malformation, syringomyelia, and rarely, hydrocephalus [Gripp et al 2015, Ejaz et al 2016, Cuoco et al 2020].
Craniofacial features include widely spaced eyes with downslanted palpebral fissures, highly arched eyebrows, ptosis, malar flattening, long philtrum, thin vermilion of the upper lip, high and narrow palate, micrognathia, and angulated ears (). Cleft palate, dental crowding, epicanthal folds, dolichocephaly, and coarse hair with a low posterior hairline can also be seen.
A high nasal voice is noted in many individuals [Gripp et al 2015].
Developmental delay, particularly gross motor delay, is frequently seen in individuals with NOTCH3-related LMS, but cognition is typically preserved. Of ten individuals with NOTCH3-related LMS, all had developmental delay; only one also had intellectual disability [Gripp et al 2015, Ejaz et al 2016, Yamada et al 2022].
Musculoskeletal. Overlap with features of connective tissue disorders include neonatal hypotonia, abdominal hernias, ligamentous laxity, keloid scars, and back pain in later life.
Many individuals have skeletal changes including scoliosis, kyphosis, vertebral fusion, scalloping of vertebrae, and wormian bones [Gripp et al 2015, Cappuccio et al 2020].
Cardiovascular concerns have included atrial septal defect, ventricular septal defect, bicuspid aortic valve, dilatation of the aorta, and coarctation of the aortic arch [Gripp et al 2015, Ejaz et al 2016, Cappuccio et al 2020].
Genitourinary. Cryptorchidism is frequently seen. Prenatal hydronephrosis with postnatal left renal hypoplasia and bilateral renal cysts was reported in one individual [Cappuccio et al 2020].
Gastrointestinal. Infants with NOTCH3-related LMS may demonstrate feeding difficulties with poor weight gain. Feeding difficulties were severe enough to warrant gastrostomy tube feeding in two individuals [Ejaz et al 2016, Brown et al 2017].
Eye abnormalities can include proptosis and oculomotor restriction.
Hearing loss. Mixed or conductive hearing loss has been noted. Brain MRI of one individual showed an apical turn of the cochlea and modiolus, and dysmorphic vestibule [Cappuccio et al 2020]