Summary
Clinical characteristics.
Neuroferritinopathy is an adult-onset progressive movement disorder characterized by chorea or dystonia and speech and swallowing deficits. The movement disorder typically affects one or two limbs and progresses to become more generalized within 20 years of disease onset. When present, asymmetry in the movement abnormalities remains throughout the course of the disorder. Most individuals develop a characteristic orofacial action-specific dystonia related to speech that leads to dysarthrophonia. Frontalis overactivity and orolingual dyskinesia are common. Cognitive deficits and behavioral issues become major problems with time.
Diagnosis/testing.
The diagnosis of neuroferritinopathy is established in a proband with typical clinical findings and/or identification of a heterozygous pathogenic variant in FTL by molecular testing.
Management.
Treatment of manifestations: While the movement disorder is particularly resistant to conventional therapy, some response has been recorded with levodopa, tetrabenazine, orphenadrine, benzhexol, sulpiride, diazepam, clonazepam, and deanol in standard doses. Botulinum toxin may be helpful for painful focal dystonia. Physical therapy is recommended to maintain mobility and prevent contractures.
Agents/circumstances to avoid: Iron supplements.
Genetic counseling.
Neuroferritinopathy is inherited in an autosomal dominant manner with 100% penetrance. Most individuals diagnosed with neuroferritinopathy have an affected parent; the proportion of individuals with neuroferritinopathy caused by a de novo pathogenic variant is unknown but likely rare. Each child of an individual with neuroferritinopathy has a 50% chance of inheriting the pathogenic variant. Prenatal and preimplantation genetic testing are possible if the FTL pathogenic variant in the family is known.
Diagnosis
No consensus clinical diagnostic criteria for neuroferritinopathy have been published.
Suggestive Findings
Neuroferritinopathy should be suspected in individuals with the following clinical, imaging, and family history findings.
Clinical findings. Adult-onset progressive movement disorder (either chorea or dystonia)
Imaging findings. Evidence of excess iron storage or accumulation on T2-weighted images from disease onset. As disease progresses, high signal on T2-weighted MRI in the caudate, globus pallidus, putamen, substantia nigra, and red nuclei is seen, followed by cystic degeneration in the caudate and putamen (see Figure 1).
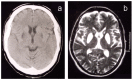
Figure 1
a. Non-contrast brain CT showing symmetric low signal in the putamina b. T2-weighted MRI image showing cystic change involving the putamina and globus pallidi and with increased signal in the heads of the caudate nuclei [Crompton et al 2005]
Family history is consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of neuroferritinopathy can be established in a proband with typical suggestive findings and/or the identification of a heterozygous pathogenic (or likely pathogenic) variant in FTL by molecular genetic testing (see Table 1).
Clinical diagnosis. Neuroferritinopathy is an adult-onset movement disorder with characteristic brain MRI findings consistent with excess brain iron deposition in the basal ganglia and an autosomal dominant family history [McNeill et al 2008]. No clinical diagnostic criteria have been published to date.
Molecular diagnosis. The molecular diagnosis of neuroferritinopathy is established in a proband with suggestive findings and identification of a heterozygous pathogenic (or likely pathogenic) variant in FTL by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variant" in this section is understood to include any likely pathogenic variant. (2) Identification of a heterozygous FTL variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with abnormal movements or neurodegenerative features are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the clinical and brain imaging findings suggest the diagnosis of neuroferritinopathy, molecular genetic testing approaches can include single-gene testing or use of a multigene panel.
- Single-gene testing. Sequence analysis of FTL to detect missense, nonsense, and splice site variants as well as small intragenic deletions/duplications. Note: Neuroferritinopathy occurs through a likely gain-of-function mechanism. Large intragenic deletions or duplications are not expected to cause neuroferritinopathy (see Genetically Related Disorders).
- A multigene panel that includes FTL and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by abnormal movements or neurodegenerative features more broadly, comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Neuroferritinopathy
Clinical Characteristics
Clinical Description
To date, more than 100 individuals have been identified with a pathogenic variant in FTL. The following description of the phenotypic features associated with this condition is based on these reports [Curtis et al 2001, Wills et al 2002, Chinnery et al 2003, Maciel et al 2005, Mancuso et al 2005, Mir et al 2005, Chinnery et al 2007, Ohta et al 2008, Devos et al 2009, Kubota et al 2009, Batey et al 2010, Ondo et al 2010, Cassidy et al 2011, Shah et al 2012, Fatima et al 2013, Storti et al 2013, Moutton et al 2014, Nishida et al 2014, Maccarinelli et al 2015, Brugger et al 2016, Ni et al 2016, Yoon et al 2019].
Table 2.
Neuroferritinopathy: Frequency of Select Features
Movement disorder. The two presenting phenotypes are typically chorea or dystonia affecting one or two limbs, although one individual presented with late-onset parkinsonism [Curtis et al 2001, Burn & Chinnery 2006, Chinnery et al 2007] and two families with cerebellar features [Vidal et al 2004, Devos et al 2009] (see Table 2). The age of onset of movement abnormalities is during adulthood (mean: 40 years, range: second to seventh decade) [Chinnery et al 2007].
- Unusual presentations have also been described, related to an underlying dystonic gait [Keogh et al 2011, Nishida et al 2014].
- The movement disorder is progressive, involving additional limbs in five to ten years and becoming more generalized within 20 years, and may lead to becoming wheelchair bound [Crompton et al 2005].
- Some individuals have striking asymmetry, which remains throughout the course of the disorder.
Oral motor manifestations
- Most individuals develop a characteristic orofacial action-specific dystonia related to speech and leading to dysarthrophonia.
- Frontalis overactivity is common, as is orolingual dyskinesia [Crompton et al 2005].
Cognitive deficits
- Cognitive issues are usually subtle but progressively worsen through the course of the disease [Crompton et al 2005]. These issues most commonly include impaired verbal fluency and verbal learning and impaired executive function [Keogh et al 2013].
- Formal neuropsychological evaluation reveals frontal/subcortical deficits that are not as prominent as those seen in Huntington disease [Wills et al 2002].
Behavioral issues. Emotional lability and aggression have been reported [Keogh et al 2013].
Prognosis. To date, there is limited information regarding prognosis in individuals with neuroferritinopathy. However, demise has been reported in individuals in mid-to-late adult life.
Neuroimaging. From disease onset, all affected individuals have evidence of excess brain iron accumulation on T2-weighted MRI. The iron deposition may be missed on other MR sequences in early stages of the disease. Later stages are associated with high signal on T2-weighted MRI in the caudate, globus pallidus, putamen, substantia nigra, and red nuclei, followed by cystic degeneration in the caudate and putamen. Neuroferritinopathy has a characteristic appearance, distinguishing it from other disorders associated with brain iron accumulation [McNeill et al 2008] and associated with progressive iron accumulation on MRI [McNeill et al 2012], including the "eye of the tiger" sign [McNeill et al 2012] and other radiologic features [Batla et la 2015].
Histopathologic examination. Of three individuals with the pathogenic FTL variant c.460dupA, histopathologic examination confirmed evidence of abnormal iron accumulation throughout the brain and particularly in the basal ganglia [Hautot et al 2007]. Affected regions contain iron and ferritin-positive spherical inclusions, often colocalizing with microglia, oligodendrocytes, and neurons. Axonal swellings (neuroaxonal spheroids) that were immunoreactive to ubiquitin, tau, and neurofilaments were also present. Mancuso et al [2005] report similar neuropathologic findings in an individual with FTL pathogenic variant c.442dupC.
Serum ferritin. Serum ferritin concentrations were low (<20 µg/L) in most males and postmenopausal females but within normal limits for premenopausal females [Chinnery et al 2007].
Genotype-Phenotype Correlations
Neuroferritinopathy-associated variants in FTL include missense variants and small intragenic duplications. Heterozygous nonsense variants are associated with L-ferritin deficiency. Deletions and heterozygous pathogenic variants in the iron-responsive element of FTL, located in the 5' UTR, are associated with hereditary hyperferritinemia cataract syndrome (see Genetically Related Disorders).
Penetrance
Penetrance is 100% [Chinnery et al 2007].
Prevalence
Prevalence is unknown. The vast majority of affected individuals described to date have the same pathogenic variant in FTL (c.460dupA; p.Arg154LysfsTer27). Evidence suggests that they have descended from a common UK founder [Chinnery et al 2003], although the identification of a person from the state of Texas with German ancestry raises the possibility of a recurrent c.460dupA pathogenic variant [Ondo et al 2010].
Genetically Related (Allelic) Disorders
Hereditary hyperferritinemia cataract syndrome (OMIM 600886). Heterozygous pathogenic variants in the iron-responsive element of FTL, located in the 5' UTR, have been identified in hereditary hyperferritinemia cataract syndrome, in which an increase in the serum ferritin concentration is associated with bilateral cataracts developing in childhood or early adult life, but not with excess iron storage in the brain. Pathogenic variants in FTL causing neuroferritinopathy are found within the coding region, and usually exon 4.
L-ferritin deficiency (OMIM 615604). Several individuals have been reported with low serum ferritin levels but without iron deficiency anemia or neurologic dysfunction. One individual was reported with a heterozygous pathogenic variant in the ATG start codon of FTL [Cremonesi et al 2004]. Another individual with a heterozygous pathogenic variant in FTL had complete absence of serum L-ferritin with otherwise normal hematologic parameters, generalized seizures, and mild neuropsychological impairment [Cozzi et al 2013].
An additional family of four individuals was reported with hypoferritinemia but no neurologic features have been described. Affected individuals in this family had a deletion involving exons 3 and 4 of FTL [Turner et al 2021].
Differential Diagnosis
Table 3.
Disorders of Interest in the Differential Diagnosis of Neuroferritinopathy
Neurodegenerative disorders with brain iron accumulation (NBIA). Neuroferritinopathy shares similar MRI appearances and clinical presentation of several other NBIAs. However, the age of onset, inheritance pattern, and T2*-weighted MRI results can be used to distinguish these disorders [McNeill et al 2008]. See Neurodegeneration with Brain Iron Accumulation Disorders Overview.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with neuroferritinopathy, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Neuroferritinopathy
Treatment of Manifestations
Table 5.
Treatment of Manifestations in Individuals with Neuroferritinopathy
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the following evaluations are recommended.
Table 6.
Recommended Surveillance for Individuals with Neuroferritinopathy
Agents/Circumstances to Avoid
Iron supplements are not recommended for affected individuals and those at risk [Chinnery et al 2007]. Iron replacement therapy with careful monitoring may be required if affected individuals develop coincidental iron deficiency anemia. There is no evidence to support avoidance of iron-rich foods by affected individuals [Author, personal observation].
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
It has been proposed that administration of deferiprone, an orally administered bidentate iron chelator that passes through the blood-brain barrier, might confer clinical and neuroradiologic improvement in individuals with neuroferritinopathy [Romano et al 2022].
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Neuroferritinopathy is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Most individuals diagnosed with neuroferritinopathy have an affected parent.
- A proband with neuroferritinopathy may have the disorder as the result of a de novo pathogenic variant. The proportion of individuals with neuroferritinopathy caused by a de novo pathogenic variant is unknown.
- If the proband appears to be the only affected family member (i.e., a simplex case), recommendations for the parents of the proband include:
- Brain MRI and measurement of serum ferritin concentration;
- Molecular genetic testing (if the FTL pathogenic variant has been identified in the proband) to confirm the genetic status of the parents and to allow reliable recurrence risk counseling.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
- The family history of some individuals diagnosed with neuroferritinopathy may appear to be negative because of failure to recognize the disorder in family members, early death of the parent before the onset of symptoms, or late onset of the disease in the affected parent. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has demonstrated that neither parent is heterozygous for the pathogenic variant identified in the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs is 50%. The penetrance of neuroferritinopathy is 100%; however, there may be differences in the age of onset and rate of progression in heterozygous sibs.
- If the proband has a known FTL pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
- If the parents have not been tested for the FTL pathogenic variant but are clinically unaffected, the sibs of a proband are still at increased risk for neuroferritinopathy because of the possibility of late onset of neuroferritinopathy in a heterozygous parent or the theoretic possibility of parental germline mosaicism.
Offspring of a proband
- Each child of an individual with neuroferritinopathy has a 50% chance of inheriting the FTL pathogenic variant.
- There may be differences in the age of onset and rate of progression of the disorder between heterozygous members of the same family.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected or has the FTL pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
Predictive testing (i.e., testing of asymptomatic at-risk individuals)
- Predictive testing for at-risk relatives is possible once the FTL pathogenic variant has been identified in an affected family member.
- Potential consequences of such testing (including, but not limited to, socioeconomic changes and the need for long-term follow up and evaluation arrangements for individuals with a positive test result) as well as the capabilities and limitations of predictive testing should be discussed in the context of formal genetic counseling prior to testing.
- Such testing is not useful in accurately predicting age of onset, severity, type of symptoms, or rate of progression in asymptomatic individuals.
Predictive testing in minors (i.e., testing of asymptomatic at-risk individuals younger than age 18 years)
- For asymptomatic minors at risk for adult-onset conditions for which early treatment would have no beneficial effect on disease morbidity and mortality, predictive genetic testing is considered inappropriate, primarily because it negates the autonomy of the child with no compelling benefit. Further, concern exists regarding the potential unhealthy adverse effects that such information may have on family dynamics, the risk of discrimination and stigmatization in the future, and the anxiety that such information may cause.
- For more information, see the National Society of Genetic Counselors position statement on genetic testing of minors for adult-onset conditions and the American Academy of Pediatrics and American College of Medical Genetics and Genomics policy statement on ethical and policy issues in genetic testing and screening of children.
In a family with an established diagnosis of neuroferritinopathy, it is appropriate to consider testing of symptomatic individuals regardless of age.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the FTL pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful. For more information, see the National Society of Genetic Counselors position statement on prenatal testing in adult-onset conditions.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- NBIA Disorders Association
- NBIAcureCenter of Excellence for NBIA Clinical Care and ResearchInternational Registry for NBIA and Related DisordersOregon Health & Science UniversityEmail: info@nbiacure.org
- Treat Iron-Related Childhood Onset Neurodegeneration (TIRCON)GermanyEmail: TIRCON@med.uni-muenchen.de
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Neuroferritinopathy: Genes and Databases
Table B.
OMIM Entries for Neuroferritinopathy (View All in OMIM)
Molecular Pathogenesis
Ferritin is an iron storage protein that has two main subunits, ferritin heavy chain (FTH) and ferritin light chain (FTL), and plays an important role in maintaining iron homeostasis. A functional ferritin molecule can store up to 4,500 iron molecules. The proportion of H and L subunits varies among tissues. FTL encodes the ferritin light chain protein. Genetic variants in FTL cause focal accumulation of iron and ferritin characteristic of neuroferritinopathy. FTL is located on chromosome 19q13.33 and consists of 4 exons and 3 introns.
The most common FTL variant, c.460dupA (p.Arg154LysfsTer27), is predicted to alter 22 C-terminal residues (the D-helix, the DE loop, and the E-helix) of the ferritin molecule, extending the protein by four amino acids. The extension is predicted to alter its iron storage capacity, possibly leading to an excessive release of toxic iron within neurons through a dominant-negative effect. Mitochondrial respiratory chain function may also be involved, as abnormal mitochondrial respiratory function has been documented in numerous individuals with neuroferritinopathy [Kurzawa-Akanbi et al 2021].
Mechanism of disease causation. Pathogenic FTL variants cause neuroferritinopathy through a gain-of-function effect.
Table 7.
Notable FTL Pathogenic Variants
Chapter Notes
Revision History
- 20 October 2022 (sw/gm) Comprehensive update posted live
- 18 January 2018 (ha) Comprehensive update posted live
- 23 December 2010 (me) Comprehensive update posted live
- 8 August 2007 (me) Comprehensive update posted live
- 30 November 2006 (pfc) Revision: sequence analysis clinically available; addition of relevant material from author's new paper, Chinnery et al [2007]
- 25 April 2005 (me) Review posted live
- 1 September 2004 (pfc) Original submission
References
Published Guidelines / Consensus Statements
- Committee on Bioethics, Committee on Genetics, and American College of Medical Genetics and Genomics Social, Ethical, Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Available online. 2013. Accessed 10-17-22.
- National Society of Genetic Counselors. Position statement on genetic testing of minors for adult-onset conditions. Available online. 2018. Accessed 10-17-22.
Literature Cited
- Batey S, Vuillaume I, Devos D, Destée A, Curtis AJ, Lombes A, Curtis A, Burn J, Chinnery PF. A novel FTL insertion causing neuroferritinopathy. J Med Genet. 2010;47:71–2. [PubMed: 20065344]
- Batla A, Adams ME, Erro R, Ganos C, Balint B, Mencacci NE, Bhatia KP. Cortical pencil lining in neuroferritinopathy: a diagnostic clue. Neurology. 2015;84:1816–8. [PMC free article: PMC4424124] [PubMed: 25832658]
- Brugger F, Kägi G, Pandolfo M, Mencacci NE, Batla A, Wiethoff S, Bhatia KP. Neurodegeneration with brain iron accumulation (NBIA) syndromes presenting with late-onset craniocervical dystonia: an illustrative case series. Mov Disord Clin Pract. 2016;4:254–7. [PMC free article: PMC6353318] [PubMed: 30838262]
- Burn J, Chinnery PF. Neuroferritinopathy. Semin Pediatr Neurol. 2006;13:176–81. [PubMed: 17101456]
- Capalbo A, Valero RA, Jimenez-Almazan J, Pardo PM, Fabiani M, Jiménez D, Simon C, Rodriguez JM. Optimizing clinical exome design and parallel gene-testing for recessive genetic conditions in preconception carrier screening: Translational research genomic data from 14,125 exomes. PLoS Genet. 2019;15:e1008409. [PMC free article: PMC6797235] [PubMed: 31589614]
- Cassidy AJ, Williams ER, Goldsmith P, Baker SN, Baker MR. The man who could not walk backward: an unusual presentation of neuroferritinopathy. Mov Disord. 2011;26:362–4. [PMC free article: PMC3060939] [PubMed: 21294155]
- Chinnery PF, Crompton DE, Birchall D, Jackson MJ, Coulthard A, Lombes A, Quinn N, Wills A, Fletcher N, Mottershead JP, Cooper P, Kellett M, Bates D, Burn J. Clinical features and natural history of neuroferritinopathy caused by the FTL1 460InsA mutation. Brain. 2007;130:110–9. [PubMed: 17142829]
- Chinnery PF, Curtis AR, Fey C, Coulthard A, Crompton D, Curtis A, Lombes A, Burn J. Neuroferritinopathy in a French family with late onset dominant dystonia. J Med Genet. 2003;40:e69. [PMC free article: PMC1735466] [PubMed: 12746423]
- Cozzi A, Santambrogio P, Privitera D, Broccoli V, Rotundo LI, Garavaglia B, Benz R, Altamura S, Goede JS, Muckenthaler MU, Levi S. Human L-ferritin deficiency is characterized by idiopathic generalized seizures and atypical restless leg syndrome. J Exp Med. 2013;210:1779–91. [PMC free article: PMC3754865] [PubMed: 23940258]
- Cremonesi L, Cozzi A, Girelli D, Ferrari F, Fermo I, Foglieni B, Levi S, Bozzini C, Camparini M, Ferrari M, Arosio P. Case report: a subject with a mutation in the ATG start codon of L-ferritin has no haematological or neurological symptoms. J Med Genet. 2004;41:e81. [PMC free article: PMC1735816] [PubMed: 15173247]
- Crompton DE, Chinnery PF, Bates D, Walls TJ, Jackson MJ, Curtis AJ, Burn J. Spectrum of movement disorders in neuroferritinopathy. Mov Disord. 2005;20:95–9. [PubMed: 15390132]
- Curtis AR, Fey C, Morris CM, Bindoff LA, Ince PG, Chinnery PF, Coulthard A, Jackson MJ, Jackson AP, McHale DP, Hay D, Barker WA, Markham AF, Bates D, Curtis A, Burn J. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001;28:350–4. [PubMed: 11438811]
- Devos D, Tchofo PJ, Vuillaume I, Destée A, Batey S, Burn J, Chinnery PF. Clinical features and natural history of neuroferritinopathy caused by the 458dupA FTL mutation. Brain. 2009;132:e109. [PMC free article: PMC2685918] [PubMed: 18854324]
- Fatima Z, Ishigame K, Araki T. Case 193: neuroferritinopathy--a brain iron accumulation and neurodegenerative disorder. Radiology. 2013;267:650–5. [PubMed: 23610097]
- Hautot D, Pankhurst QA, Morris CM, Curtis A, Burn J, Dobson J. Preliminary observation of elevated levels of nanocrystalline iron oxide in the basal ganglia of neuroferritinopathy patients. Biochim Biophys Acta. 2007;1772:21–5. [PMC free article: PMC1993816] [PubMed: 17097860]
- Keogh MJ, Khan A, Gorman G, McNeill A, Horvath R, Burn J, Chinnery PF. An unusual gait following the discovery of a new disease. Pract Neurol. 2011;11:81–4. [PubMed: 21385964]
- Keogh MJ, Singh B, Chinnery PF. Early neuropsychiatry features in neuroferritinopathy. Mov Disord. 2013;28:1310–3. [PubMed: 23436236]
- Kubota A, Hida A, Ichikawa Y, Momose Y, Goto J, Igeta Y, Hashida H, Yoshida K, Ikeda S, Kanazawa I, Tsuji S. A novel ferritin light chain gene mutation in a Japanese family with neuroferritinopathy: description of clinical features and implications for genotype-phenotype correlations. Mov Disord. 2009;24:441–5. [PubMed: 19117339]
- Kurzawa-Akanbi M, Keogh M, Tsefou E, Ramsay L, Johnson M, Keers S, Wsa Ochieng L, McNair A, Singh P, Khan A, Pyle A, Hudson G, Ince PG, Attems J, Burn J, Chinnery PF, Morris CM. Neuropathological and biochemical investigation of Hereditary Ferritinopathy cases with ferritin light chain mutation: Prominent protein aggregation in the absence of major mitochondrial or oxidative stress. Neuropathol Appl Neurobiol. 2021;47:26–42. [PubMed: 32464705]
- Maccarinelli F, Pagani A, Cozzi A, Codazzi F, Di Giacomo G, Capoccia S, Rapino S, Finazzi D, Politi LS, Cirulli F, Giorgio M, Cremona O, Grohovaz F, Levi S. A novel neuroferritinopathy mouse model (FTL 498InsTC) shows progressive brain iron dysregulation, morphological signs of early neurodegeneration and motor coordination deficits. Neurobiol Dis. 2015;81:119–33. [PMC free article: PMC4642750] [PubMed: 25447222]
- Maciel P, Cruz VT, Constante M, Iniesta I, Costa MC, Gallati S, Sousa N, Sequeiros J, Coutinho P, Santos MM. Neuroferritinopathy: missense mutation in FTL causing early-onset bilateral pallidal involvement. Neurology. 2005;65:603–5. [PMC free article: PMC2886026] [PubMed: 16116125]
- Mancuso M, Davidzon G, Kurlan RM, Tawil R, Bonilla E, Di Mauro S, Powers JM. Hereditary ferritinopathy: a novel mutation, its cellular pathology, and pathogenetic insights. J Neuropathol Exp Neurol. 2005;64:280–94. [PubMed: 15835264]
- Marchand F, Moreau C, Kuchcinski G, Huin V, Defebvre L, Devos D. Conservative iron chelation for neuroferritinopathy. Mov Disord. 2022;37:1948–52. [PMC free article: PMC10360136] [PubMed: 35996824]
- McNeill A, Birchall D, Hayflick SJ, Gregory A, Schenk JF, Zimmerman EA, Shang H, Miyajima H, Chinnery PF. T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation. Neurology. 2008;70:1614–9. [PMC free article: PMC2706154] [PubMed: 18443312]
- McNeill A, Gorman G, Khan A, Horvath R, Blamire AM, Chinnery PF. Progressive brain iron accumulation in neuroferritinopathy measured by the thalamic T2* relaxation rate. AJNR Am J Neuroradiol. 2012;33:1810–3. [PMC free article: PMC4038493] [PubMed: 22499840]
- Mir P, Edwards MJ, Curtis AR, Bhatia KP, Quinn NP. Adult-onset generalized dystonia due to a mutation in the neuroferritinopathy gene. Mov Disord. 2005;20:243–5. [PubMed: 15390032]
- Moutton S, Fergelot P, Trocello JM, Plante-Bordeneuve V, Houcinat N, Wenisch E, Larue V, Brugières P, Clot F, Lacombe D, Arveiler B, Goizet C. A novel FTL mutation responsible for neuroferritinopathy with asymmetric clinical features and brain anomalies. Parkinsonism Relat Disord. 2014;20:935–7. [PubMed: 24907184]
- Ni W, Li HF, Zheng YC, Wu ZY. FTL mutation in a Chinese pedigree with neuroferritinopathy. Neurol Genet. 2016;2:e74. [PMC free article: PMC4851275] [PubMed: 27158664]
- Nishida K, Garringer HJ, Futamura N, Funakawa I, Jinnai K, Vidal R, Takao M. A novel ferritin light chain mutation in neuroferritinopathy with an atypical presentation. J Neurol Sci. 2014;342:173–7. [PMC free article: PMC4048789] [PubMed: 24825732]
- Ohta E, Nagasaka T, Shindo K, Toma S, Nagasaka K, Ohta K, Shiozawa Z. Neuroferritinopathy in a Japanese family with a duplication in the ferritin light chain gene. Neurology. 2008;70:1493–4. [PubMed: 18413574]
- Ondo WG, Adam OR, Jankovic J, Chinnery PF. Dramatic response of facial stereotype/tic to tetrabenazine in the first reported cases of neuroferritinopathy in the United States. Mov Disord. 2010;25:2470–2. [PubMed: 20818611]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Romano N, Baiardi G, Pinto VM, Quintino S, Gianesin B, Sasso R, Diociasi A, Mattioli F, Marchese R, Abbruzzese G, Castaldi A, Forni GL. Long-term neuroradiological and clinical evaluation of NBIA patients treated with a deferiprone based iron-chelation therapy. J Clin Med. 2022;11:4524. [PMC free article: PMC9369383] [PubMed: 35956138]
- Shah SO, Mehta H, Fekete R. Late-onset neurodegeneration with brain iron accumulation with diffusion tensor magnetic resonance imaging. Case Rep Neurol. 2012;4:216–23. [PMC free article: PMC3531933] [PubMed: 23275784]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Storti E, Cortese F, Di Fabio R, Fiorillo C, Pierallini A, Tessa A, Valleriani A, Pierelli F, Santorelli FM, Casali C. De novo FTL mutation: a clinical, neuroimaging, and molecular study. Mov Disord. 2013;28:252–3. [PubMed: 23239021]
- Turner S, Dress C, Misra VK. A. 3'-truncating FTL mutation associated with hypoferritinemia without neuroferritinopathy. Eur J Med Genet. 2021;64:104159. [PubMed: 33548513]
- Vidal R, Ghetti B, Takao M, Brefel-Courbon C, Uro-Coste E, Glazier BS, Siani V, Benson MD, Calvas P, Miravalle L, Rascol O, Delisle MB. Intracellular ferritin accumulation in neural and extraneural tissue characterizes a neurodegenerative disease associated with a mutation in the ferritin light polypeptide gene. J Neuropathol Exp Neurol. 2004;63:363–80. [PubMed: 15099026]
- Wills AJ, Sawle GV, Guilbert PR, Curtis AR. Palatal tremor and cognitive decline in neuroferritinopathy. J Neurol Neurosurg Psychiatry. 2002;73:91–2. [PMC free article: PMC1757327] [PubMed: 12082064]
- Yoon SH, Kim NY, Kim YJ, Lyoo CH. Novel ferritin light chain gene mutation in a Korean patient with neuroferritinopathy. J Mov Disord. 2019;12:63–5. [PMC free article: PMC6369382] [PubMed: 30732435]
Publication Details
Author Information and Affiliations
University of Cambridge
Cambridge, United Kingdom
Publication History
Initial Posting: April 25, 2005; Last Update: October 20, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Chinnery PF. Neuroferritinopathy. 2005 Apr 25 [Updated 2022 Oct 20]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
