Summary
Clinical characteristics.
NDP-related retinopathies typically involve bilateral and symmetric fibrovascular changes of the retina that are evident at birth and usually progress through childhood or adolescence to cause varying degrees of visual impairment. The spectrum of NDP-related retinopathies appears to be a continuum with considerable overlap, ranging from Norrie disease, NDP-related persistent fetal vasculature, NDP-related familial exudative vitreoretinopathy, NDP-related advanced retinopathy of prematurity, and NDP-related Coats disease. The eye findings of Norrie disease, the first described and best characterized of these disorders, are typically bilateral grayish-yellow, glistening, elevated retrolental masses composed of immature retinal cells usually visible through clear lenses; foveal development is always incomplete. In addition, Norrie disease is the only NDP-related retinopathy with associated extraocular findings, which can variably include cognitive disability, mental health and behavior disorders, seizures, sensorineural hearing loss, and peripheral vascular disease.
Rarely, females who are heterozygous for an NDP pathogenic variant have clinical manifestations including retinal and extraocular features that may vary between other family members heterozygous for the same NDP pathogenic variant.
Diagnosis/testing.
The diagnosis of an NDP-related retinopathy, an X-linked disorder, is established in a male proband with suggestive clinical findings by identification on molecular genetic testing of a hemizygous pathogenic variant in NDP; and in a female proband with suggestive clinical findings by identification of a heterozygous pathogenic variant in NDP.
Management.
Treatment of manifestations: Based on a child's ocular findings, pediatric ophthalmologists and pediatric vitreoretinal surgeons individualize management to reduce increased intraocular pressure to decrease the risk of glaucoma; treat refractive errors and/or strabismus to optimize vision and ocular alignment; and discuss risks and benefits of possible vision-saving surgical intervention. Other issues are addressed by multidisciplinary specialists, typically: low-vision specialists, neurologists, pediatricians, developmental pediatricians, pediatric sleep specialists, mental health specialists, physiatrists, occupational and physical therapists, audiologists, and speech-language pathologists.
Surveillance: Routine monitoring of: ophthalmologic manifestations and low vision needs; hearing; developmental progress and educational needs; neurologic and mental health manifestations; peripheral vascular disease; and response to treatment.
Agents/circumstances to avoid: Situations that could exacerbate hearing loss such as loud noises and use of listening devices that amplify sound.
Evaluation of relatives at risk: Clarify the genetic status of apparently asymptomatic older and younger at-risk female relatives so that those with the familial NDP pathogenic have the option to undergo fluorescein angiography to detect peripheral retinal vascular changes that may require close monitoring and/or prophylactic laser photocoagulation to decrease the risk of neovascularization and vitreoretinal traction.
Genetic counseling.
NDP-related retinopathies are inherited in an X-linked manner. The majority of affected males have the disorder as the result of an NDP pathogenic variant inherited from their mother. If the mother of the proband has an NDP pathogenic variant (i.e., is a carrier), the chance of transmitting it in each pregnancy is 50%: males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be heterozygous and may rarely have clinical manifestations. Affected males transmit the NDP pathogenic variant to all of their daughters and none of their sons. Once the NDP pathogenic variant has been identified in an affected family member, testing for females at risk of being carriers and prenatal and preimplantation genetic testing are possible.
GeneReview Scope
Table
Norrie disease (classic Norrie disease ocular phenotype with or without extraocular findings) NDP-related persistent fetal vasculature (PFV)
Diagnosis
Suggestive Findings
An NDP-related retinopathy should be suspected in a male infant with the following ocular findings and family history.
Ocular findings. The following spectrum of typically bilateral and symmetric fibrovascular changes of the retina are evident at birth and usually progress through childhood or adolescence to cause varying degrees of visual impairment:
- Norrie disease
- Retrolental grayish-yellow fibrovascular masses composed of immature retinal cells (pseudogliomas) secondary to retinal vascular dysgenesis
- Retinal detachment
- Cataract
- Infantile blindness
- NDP-related persistent fetal vasculature (PFV; formerly referred to as persistent hyperplastic primary vitreous [PHPV]). Fibrotic white stalk with hyaloid vessel remnants extending from optic disc to posterior lens capsule with or without cataract
- NDP-related bilateral familial exudative vitreoretinopathy (FEVR). Peripheral retinal avascularity with or without congenital retinal folds, temporal dragging of the macula, retinal neovascularization, and fibrovascular scarring at the ora serrata
- NDP-related advanced retinopathy of prematurity (ROP). Fibrovascular proliferation, end-stage retrolental fibroplasia, retinal detachment secondary to vitreoretinal traction, and retinal neovascularization
- NDP-related Coats disease. Unilateral retinal telangiectasia, exudation with or without retinal detachment, and subretinal fibrosis
Family history is consistent with X-linked inheritance (e.g., no male-to-male transmission). Absence of a known family history does not preclude the diagnosis (see Genetic Counseling).
Establishing the Diagnosis
The diagnosis of an NDP-related retinopathy is established in a male proband with suggestive clinical findings by identification of a hemizygous pathogenic (or likely pathogenic) variant in NDP on molecular genetic testing, and in a female proband with suggestive findings and a heterozygous pathogenic (or likely pathogenic) variant in NDP identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a hemizygous NDP variant of uncertain significance does not establish or rule out the diagnosis of an NDP-related retinopathy.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) with consideration of chromosome microarray to establish breakpoints in the event of a whole-gene deletion.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1), whereas genomic testing does not (see Option 2).
Option 1
When the phenotypic findings and family history suggest the diagnosis of an NDP-related retinopathy, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of NDP is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected.If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
- A multigene panel that includes NDP and other genes associated with overlapping ocular phenotypes (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene(s) are likely involved. Exome sequencing is most commonly used; genome sequencing is becoming more available. Note: Unlike exome sequencing, genome sequencing can identify variants outside of the coding region. Although most confirmed pathogenic variants identified by genome sequencing are within exons [Taylor et al 2015], a pathogenic variant has been detected in the noncoding region of NDP [Daich Varela et al 2023].
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in NDP-Related Retinopathies
Clinical Characteristics
Clinical Description
NDP-related ocular phenotypes typically involve bilateral and symmetric fibrovascular changes of the retina that are evident at birth and usually progress through childhood or adolescence to cause varying degrees of visual impairment. The NDP-related ocular phenotypes appear to be a continuum with considerable overlap: Norrie disease, NDP-related persistent fetal vasculature (PFV), NDP-related familial exudative vitreoretinopathy (FEVR), NDP-related advanced retinopathy of prematurity (ROP), and NDP-related Coats disease [Black et al 1999, Dickinson et al 2006] (Table 2).
Table 2.
NDP-Related Retinopathies: Ocular Phenotypes
Norrie Disease
Ocular findings. The ocular findings in Norrie disease are the first described and best characterized eye findings of the NDP-related retinopathies.
In newborns and infants, the classic finding is a grayish-yellow, glistening, elevated retrolental mass composed of immature retinal cells that is usually visible through a clear lens in both eyes. These masses are referred to as "pseudogliomas" because they resemble tumors such as retinoblastoma. There is retinal dysplasia and incomplete foveal development in all cases. Partial or complete retinal detachments are often present at birth in both eyes. If not present at birth, retinal detachments may evolve over the first few months of life.
Nystagmus is common secondary to profound, bilateral vision loss [Smith et al 2012].
The irides, anterior chambers, and corneal diameter may be abnormal in size and appearance as a result of anterior segment dysgenesis.
The size of the globe may be normal, smaller (microphthalmia), or enlarged (buphthalmos).
Intraocular pressure is often normal at birth but can become elevated (i.e., secondary glaucoma) as a result of malformations of the anterior chamber and angle resulting in impaired outflow through the trabecular meshwork. Other consequences of impaired outflow include buphthalmos, pain, and further progression of vision loss.
From infancy through childhood, progressive changes typically include opacification of the lens (cataract) and cornea, atrophy or hypoplasia of the iris with adhesions forming between the lens and the iris (posterior synechiae) and between the iris and the cornea (anterior synechiae), and shallowing of the anterior chamber. Hemorrhagic necrosis of the undifferentiated retinal masses can occur.
These changes are followed by corneal opacification (e.g., band keratopathy), loss of intraocular pressure (hypotony), and shrinkage of the globe (phthisis bulbi) usually within the first decade of life. In the end stage of the Norrie disease ocular phenotype, the corneas appear milky and opacified, and the globes appear small and sunken in the orbits [Drenser et al 2007].
The majority of affected males lose all light perception during the first year of life.
Extraocular findings
- Cognitive disability. In a large series, 14 of 51 of males with the Norrie disease phenotype had developmental delay / intellectual disability [Smith et al 2012].
- Mental health and behavior. While most affected individuals are cognitively normal and develop healthy relationships, nearly all report a period of depression coincident with the onset of hearing loss. For individuals who are blind, becoming deaf-blind results in increased social isolation. Autism spectrum disorder has been reported in one third of affected individuals. Emotional lability has been reported in up to one fourth of affected individuals. In one large series, only one individual was reported with mental illness [Smith et al 2012].
- Seizures have been reported in 16 of 56 individuals with Norrie disease. Spontaneous resolution of seizures was reported in 50% [Smith et al 2012].
- Auditory findings. Hearing initially waxes and wanes, followed by slow deterioration over time. By the late teenage years, 42 of 56 males with Norrie disease in one report had hearing loss, with an increase to 80%-90% by their late 20s (Figure 1) [Smith et al 2012].In a report of six affected males, onset of hearing loss ranged between ages three and 35 years [Bryant et al 2022]. Audiograms showed significant fluctuation: one individual had an abnormal audiogram at age eight years, a normal audiogram at age nine years, and subsequently abnormal audiograms at age 10 years and thereafter.Speech discrimination is relatively well preserved even when the threshold hearing loss is severe [Halpin et al 2005, Halpin & Sims 2008]. Ear fullness ("stuffiness") and tinnitus were also reported [Smith et al 2012].Hearing loss typically progresses over time. Three individuals whose hearing loss progressed over time reported significantly improved quality of life after cochlear implantation [Smith et al 2012, Bryant et al 2022].
- Peripheral vascular disease (Figure 2) includes the following [Michaelides et al 2004, Smith et al 2012]:
- Varicosities in the lower extremities followed by development of stasis ulcers have been reported in nearly half of all affected males more than 16 years old.
- Erectile dysfunction has been reported in 14 of 20 adults.
- Intra- and interfamilial phenotypic variability has been noted in the Norrie disease retinal phenotype [Wu et al 2007] and its extraocular manifestations [Khan et al 2004, Allen et al 2006]. Although variable intellectual disability was noted in one of two brothers [Zhang et al 2013], in other reports of multiple brothers with Norrie disease all had intellectual disability [Arai et al 2014].
- Life span may be shortened by general risks associated with intellectual disability, blindness, and/or hearing loss, such as increased risk of trauma, complications related to a seizure disorder, and peripheral vascular disease.
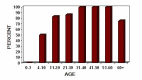
Figure 1.
Hearing loss by age group in a subset of affected males enrolled in the Norrie Disease Registry (n=56) [Smith et al 2012]
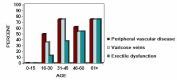
Figure 2.
Peripheral vascular disease by age group in a subset of affected males enrolled in the Norrie Disease Registry (n=56) [Smith et al 2012]
NDP-Related Persistent Fetal Vasculature (PFV)
Formerly referred to as persistent hyperplastic primary vitreous (PHPV), PFV is characterized by a fibrotic white stalk with hyaloid vessel remnants extending from the optic disc to the posterior lens capsule. NDP-related PFV may be unilateral or bilateral. The retina may be in folds or detached as a result of vitreoretinal traction from the stalk. The lens may or may not be clear.
If a stalk persists without surgical intervention, restricted growth of the globe through childhood can lead to further vitreoretinal traction, retinal exudation, retinal detachment, secondary glaucoma, and/or phthisis bulbi. The anterior segment, pars plana, and vitreous base are often malformed with anteriorization of retinal tissue past the ora serrata, which increases the complexity of surgical procedures. Although progression to complete retinal detachment has been described, it is not clear if such progression always occurs [Wu et al 2007].
NDP-Related X-Linked Familial Exudative Vitreoretinopathy (FEVR)
NDP-related X-linked FEVR is characterized by premature arrest of vascularization of the retina resulting in peripheral retina avascularity of both eyes. This avascular zone may be the only retinal finding; however, when more severe, congenital falciform retinal folds are present, and the macula may be dragged temporally, leading to macula displacement (i.e., macular ectopia) [Shute & McLoone 2022, Yang et al 2022].
Peripheral retinal ischemia may lead to retinal or extraretinal neovascularization. Such eye findings may progress to retinal detachment either through increasing traction on the retina from progressive fibrovascular changes in the temporal retinal periphery or through exudation of serous fluid by the fragile capillaries in the abnormal peripheral retinal vasculature. Retinal detachment can be accompanied by a decrease in central visual acuity as a result of macular involvement. Stage 5 total retinal detachments may be indistinguishable from the Norrie disease ocular phenotype; however, NDP-related FEVR is typically nonsyndromic (not associated with, e.g., hearing loss, intellectual disability, seizures).
Variable expressivity of NDP-related FEVR includes asymmetry of ophthalmologic findings and/or intrafamilial variability among families with the same NDP pathogenic variant.
NDP-Related Advanced Retinopathy of Prematurity (ROP)
NDP-related advanced ROP (Stage 4B/5) involves retinal changes similar to those found in NDP-related FEVR.
Pathogenic variants in NDP were identified in four of 16 premature infants with advanced ROP that resembled the Norrie disease ocular phenotype [Hiraoka et al 2001]. A study of 17 infants with advanced ROP identified one infant with an NDP pathogenic variant in the 5' UTR [Hiraoka et al 2010]. More recently, three of nine infants with atypical ROP had hemizygous pathogenic NDP variants; the remainder had pathogenic variants in other genes associated with FEVR [Li et al 2020].
NDP-Related Coats Disease
NDP-related Coats disease is an exudative proliferative vasculopathy with onset usually in infancy or childhood, and typically before age 20 years. Retinal vascular changes include telangiectasias, venous and capillary fusiform dilatation, and microaneurysms. Subretinal lipid exudate and retinal hemorrhage are observed, usually in the macula and/or superotemporal regions. Exudative retinal detachment and decreased retinal capillary perfusion may occur. Other complications can include iridocyclitis, cataract, and/or neovascular glaucoma. One woman with NDP-related Coats disease had a son with the Norrie disease ocular phenotype [Black et al 1999].
Heterozygous Females
Clinical manifestations in heterozygous females are rare and usually presumed to be secondary to non-random (unfavorable) X-chromosome inactivation. NDP-related ocular manifestations in heterozygous females have included:
- Retinal detachment, peripheral retinal avascularity, neovascularization, exudation, and high hyperopia [Lin et al 2010, Parzefall et al 2014], and associated vision loss [Yamada et al 2001];
- Extraocular findings that include mild sensorineural hearing loss [Halpin et al 2005].
Phenotypic expression has also been reported in two women with an X-autosome translocation [Meire et al 1998].
Intrafamilial variability may be observed among heterozygous female family members. In one family segregating an NDP pathogenic variant, the maternal grandmother had Norrie disease with bilateral congenital blindness and progressive hearing loss, whereas a daughter heterozygous for the same NDP pathogenic variant was unaffected [Shastry et al 1999].
Genotype-Phenotype Correlations
The Norrie disease ocular phenotype, especially at birth, is more severe than that of NDP-related FEVR.
- NDP variants affecting the cysteine knot domain of the norrin protein result in Norrie disease ocular phenotypes and, possibly, phenotypic expression in heterozygous females [Wu et al 2007, Lin et al 2010, Parzefall et al 2014].
- In contrast, males with non-cysteine variants (which do not affect the tertiary structure of the protein) usually have the less severe ocular findings more consistent with NDP-related FEVR, including avascular peripheral retina, abnormal retinal vasculature, and subretinal exudation [Wu et al 2007].
Nomenclature
The term "Norrie disease" used in the literature typically refers to the classic Norrie disease ocular phenotype occurring with or without extraocular features.
Outdated names for Norrie disease include Anderson-Warburg syndrome, atrophia bulborum hereditarian, Episkopi blindness, Norrie-Warburg syndrome, and pseudoglioma congenita.
Persistent fetal vasculature (PFV) was formerly referred to as persistent hyperplastic primary vitreous (PHPV).
NDP-related familial exudative vitreoretinopathy may also be referred to as X-linked familial exudative vitreoretinopathy.
Prevalence
No incidence or prevalence figures for NDP-related retinopathies are available.
In one academic institution with a large pediatric retina practice, 109 individuals with vitreoretinopathies were enrolled in a three-year prospective study. Eleven of the 109 individuals had NDP pathogenic variants: five with NDP-related Norrie disease, one with bilateral NDP-related PFV, four with NDP-related FEVR, and one with NDP-related advanced ROP [Wu et al 2007].
Norrie disease has been reported in many populations, including northern and central European, American of European descent, African American, French Canadian, Hispanic, Chinese, Iranian [Talebi et al 2018], Indian [Ghosh et al 2012, Sudha et al 2018], and Japanese.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with intragenic germline pathogenic variants in NDP.
Xp11.3 contiguous gene deletions
- Deletions extending beyond NDP are associated with phenotypes more severe than those of intragenic NDP pathogenic variants [Suárez-Merino et al 2001, Wu et al 2007].
- Deletions including NDP as well as MAOA, MAOB, and EFHC2 may be associated with more complex and variable phenotypes ranging from isolated ocular features of congenital blindness and leukocoria to syndromic findings including intellectual disability, epilepsy, and microcephaly [Sims et al 1989, Rodriguez-Revenga et al 2007, Smith et al 2012, Jia et al 2017, Bortolato et al 2018].
Differential Diagnosis
Table 3.
Genes of Interest in the Differential Diagnosis of NDP-Related Retinopathies
Retinopathy of prematurity (ROP). The retinal findings in the Norrie disease ocular phenotype can mimic persistent fetal vasculature (PFV) and ROP. ROP – previously described as retrolental fibroplasia – typically occurs in infants with prematurity and low birth weight. Although ROP is still a leading cause of preventable childhood blindness worldwide, improved oxygen supplementation protocols in preterm neonates have resulted in a decreased incidence of ROP. Thus, bilateral advanced ROP with or without hearing loss in a preterm child should raise suspicion of a possible genetic component.
Management
No clinical practice guidelines for NDP-related retinopathies have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with an NDP-related retinopathy, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with NDP-Related Retinopathies
Treatment of Manifestations
Ocular Manifestations
Pediatric ophthalmologists and pediatric vitreoretinal surgeons must individualize management based on a child's ocular development and anatomy. Early intervention, ideally at the time of diagnosis, can be vision saving.
- Reduction of the intraocular pressure to decrease the risk of glaucoma may require use of topical or systemic agents.
- A pediatric ophthalmologist should manage refractive errors and/or strabismus to optimize vision and ocular alignment.
- A pediatric vitreoretinal surgeon, when appropriate, should discuss risks and benefits of surgical intervention. Operating on a less developed eye, especially one with retinal dysplasia and/or other abnormalities, leaves minimal room for error. Advanced vitreoretinopathy with significant retinal dysplasia may be too severe to merit surgical intervention.
The following information represents additional management recommendations, albeit generalized, for the ocular manifestations of the NDP-related retinopathies.
Norrie disease. Because the majority of individuals with Norrie disease have complete retinal detachment at birth, surgical interventions may not help preserve sight. As untreated eyes often progress to phthisis with no light perception, some vitreoretinal surgeons and parents may choose to proceed with surgery in young children with the goal to maintain eye size and/or appearance or to preserve visual function, even if limited to light perception.
Individuals with Norrie disease who do not have complete retinal detachment may benefit from surgery and/or laser therapy per the following reports:
- Laser ablation of the avascular retina in both eyes of a two-day-old infant after planned delivery at 34 weeks' gestation (following exclusion of retinal detachment by fetal ultrasonography at 28 and 33 weeks' gestation) revealed complete attachment of the retinas in both eyes at age ten months [Sisk et al 2014].Following successful prophylactic laser photocoagulation at birth at 37 weeks' gestation, Teller visual acuity testing at age 23 months revealed age-appropriate vision of 20/100 [Chow et al 2010].Thus, families may wish to consider the option of preterm genetic testing, and retinal laser treatment of an affected male immediately after birth with the intent to preserve vision.
- In a retrospective review of medical records in a tertiary care pediatric retinal clinical practice (1988-2008), seven of 14 male infants with the Norrie disease ocular phenotype who underwent vitrectomy by age 12 months (median 4.5 months) had documented maintenance of light perception in at least one eye [Walsh et al 2010].
- When translimbal iridectomy, lensectomy, capsulectomy, and vitrectomy are indicated, it is recommended that extreme caution be used to avoid intraoperative traction. When there is a total retinal detachment (Stage 5), the lens and capsule should be removed to eliminate a scaffold for anterior proliferation and to avoid iatrogenic breaks.
- Early simultaneous intervention in both eyes is feasible and safe for children with bilateral vitreoretinal involvement in order to avoid a second anesthesia [Yonekawa et al 2016].
In the progressive stage of the ocular findings of Norrie disease, development of increased intraocular pressure may require medications and/or surgical intervention.
Rarely, enucleation is required to control pain in a blind eye caused by hypotony/phthisis.
NDP-related persistent fetal vasculature (PFV). The PFV (formerly referred to as persistent hyperplastic primary vitreous [PHPV]) stalk between the retina and the lens restricts growth of the globe, often requiring surgical intervention to sever this connection. The complexity of this surgical procedure is increased when anteriorization of retinal tissue past the ora serrata has caused malformation of the anterior segment, pars plana, and vitreous base. Thus, the surgeon has to distinguish the PFV stalk from any retinal fold(s), as cutting the retinal tissue posteriorly can lead to proliferative vitreoretinopathy and/or retinal detachment.
NDP-related familial exudative vitreoretinopathy (FEVR)
- Prophylactic laser photocoagulation of avascular peripheral retina at the time of diagnosis can decrease the production of vascular endothelial growth factor (VEGF) and decrease the risk of neovascularization and vitreoretinal traction.
- Early careful surgical dissection of tractional tissues can potentially result in good anatomic outcomes with FEVR-associated retinal detachment.
NDP-related advanced retinopathy of prematurity (ROP)
- Management is similar to that for NDP-related FEVR with prophylactic laser photocoagulation of avascular peripheral retina and surgical dissection of tractional tissues when vitreoretinal traction (usually temporally) is significant.
- Severing transvitreal traction vectors should guide surgical management of extrafoveal retinal detachment (Stage 4A), subtotal foveal-involving retinal detachment (Stage 4B), and total retinal detachment (Stage 5).
- Because children born preterm have an increased prevalence of all refractive errors (especially myopia, and especially after laser treatment), these should be diagnosed and managed promptly to decrease the risk of amblyopia.
NDP-related Coats disease
- Laser photocoagulation (directed to telangiectatic vessels, microaneurysms, and avascular areas of peripheral retina) may be warranted when there is significant subretinal exudation and/or hemorrhage that affects the macula.
- Anti-VEGF agents (e.g., bevacizumab) have been increasingly used to control the exudative process in Coats disease.
Extraocular Manifestations
Treatment involves a multidisciplinary team, typically including low-vision services, neurologists, pediatricians, developmental pediatricians, pediatric sleep specialists, mental health specialists, physiatrists, occupational and physical therapists, audiologists, and speech-language pathologists (Table 5).
Table 5.
Treatment of Manifestations in Individuals with NDP-Related Retinopathies
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as special educators and low vision education specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies, and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider.
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
- State deaf-blind services:
- In addition to the educational services in the US discussed above, state-level federally funded programs are mandated to provide services for individuals from birth to age 21 years with combined hearing and vision issues (nationaldb.org). Of note, the designation "deaf-blind," used to qualify individuals with combined vision and hearing loss for these services, does not imply total hearing loss or total vision loss.
- State deaf-blind services typically provide information and training to families, technical assistance to schools and early intervention programs, and assistance with IEPs and transitions.
Sleep Issues
Abnormal sleep patterns may develop in individuals with no light perception and abnormal circadian rhythmicity. Frequent early morning waking and daytime sleeping can be mitigated with strict daily schedules and medications (e.g., melatonin), often managed by a pediatric sleep specialist or developmental pediatrician [Davitt et al 1997].
Surveillance
Table 6.
Recommended Surveillance for Individuals with NDP-Related Retinopathies
Agents/Circumstances to Avoid
Given the risk of hearing loss, the following are recommended:
- Avoidance of exposure to loud noises
- Use of hearing protection in noisy environments or when using noisy equipment
- Minimal use of ear buds and other listening devices
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk female relatives of an affected individual. Females who are heterozygous for the familial NDP pathogenic variant have the option to undergo fluorescein angiography for detection of peripheral retinal vascular changes and avascular regions that may require close monitoring and/or prophylactic laser photocoagulation to decrease production of vascular endothelial growth factor and decrease the risk of neovascularization and vitreoretinal traction.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
NDP-related retinopathies are inherited in an X-linked manner.
Risk to Family Members
Parents of a male proband
- The father of an affected male will not have the disorder nor will he be hemizygous for the NDP pathogenic variant; therefore, he does not require further evaluation/testing.
- The majority of affected males have the disorder as the result of an NDP pathogenic variant inherited from their mother. The heterozygous mother of a proband will typically not have clinical manifestations of the disorder (see Heterozygous Females).
- In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote. If a woman has more than one affected child and no other affected relatives, and if the NDP pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism.
- If a male is the only affected family member (i.e., a simplex case):
- The mother may be a heterozygote;
- The affected male may have a de novo NDP pathogenic variant, in which case the mother is not a heterozygote (de novo NDP pathogenic variants are rare);
- The mother may have somatic/germline mosaicism.
- Molecular genetic testing of the mother is recommended to confirm her genetic status, allow reliable recurrence risk assessment, and clarify her risk for NDP-related retinal disease (see Evaluation of Relatives at Risk).
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
- If the mother of the proband has an NDP pathogenic variant, the chance of transmitting it in each pregnancy is 50%:
- Males who inherit the pathogenic variant will be affected. (Note: Intrafamilial phenotypic variability may be observed in the retinal phenotype and extraocular manifestations of the disorder; see Clinical Characteristics.)
- Females who inherit the pathogenic variant will be heterozygotes. Clinical findings in heterozygous females are rare and are usually presumed to be secondary to non-random (unfavorable) X-chromosome inactivation.Expression in heterozygous females can include retinal and extraocular features and may vary between heterozygous female family members (see Heterozygous Females). In one family segregating an NDP pathogenic variant, the maternal grandmother had Norrie disease with bilateral congenital blindness and progressive hearing loss, whereas an unaffected daughter was heterozygous for the same NDP pathogenic variant [Shastry et al 1999].
- If the proband represents a simplex case and if the NDP pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is greater than that of the general population because of the possibility of maternal germline mosaicism.
Offspring of a male proband. Affected males transmit the NDP pathogenic variant to all of their daughters and none of their sons.
Other family members. The maternal aunts and maternal cousins of a male proband may be at risk of having an NDP pathogenic variant.
Heterozygote Detection
In rare instances, females heterozygous for an NDP pathogenic variant may have retinal and extraocular features (see Heterozygous Females). It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk female relatives of an affected individual in order to identify as early as possible those who would benefit from prompt initiation of treatment and preventive measures (see Management, Evaluation of Relatives at Risk).
Molecular genetic testing to identify female heterozygotes is most informative if the NDP pathogenic variant has been identified in an affected family member.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are heterozygous, or are at risk of being heterozygous.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the NDP pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for NDP-related retinopathies is possible.
Ultrasound imaging. While ultrasound imaging of an affected fetus is typically normal in the first two trimesters, ultrasound imaging may allow for detection of ophthalmic features related to the Norrie disease ocular phenotype during the third trimester of pregnancy. This has been described in a male fetus at 34 weeks exhibiting bilateral retinal detachment, with an affected sib previously diagnosed [Redmond et al 1993]; in a male fetus at 36 weeks 5 days exhibiting bilateral massive vitreous cavity opacities and retinal detachment, with a previous normal ultrasound at 31 weeks 4 days and an affected sib [Wu et al 2017]; and in a pregnancy with a negative family history of Norrie disease with ultrasound showing bilateral intraocular hyperechogenic opacities behind the lenses at 31 weeks 5 days [Dubucs et al 2019].
Consideration of prompt ophthalmologic management after birth can provide the greatest opportunity for anatomic success of retinal repair [Shima et al 2009, Chow et al 2010, Walsh et al 2010]. Because retinal detachment may occur in the late third trimester, preterm induction of labor may be considered for prophylactic surgery [Chow et al 2010].
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- Norrie Disease FoundationPO Box 12476Colchester CO1 9RBUnited Kingdom
- American Council of the Blind (ACB)2200 Wilson BoulevardSuite 650Arlington VA 22201Phone: 800-424-8666 (toll-free); 202-467-5081Fax: 202-467-5085Email: info@acb.org
- American Society for Deaf ChildrenPhone: 800-942-2732 (ASDC)Email: info@deafchildren.org
- Family ConnectFor parents of children who are blind or visually impaired.Phone: 800-232-5463Email: connectcenter@aph.org
- Genetic and Rare Diseases Information Center (GARD)Phone: 888-205-2311
- National Association of the DeafPhone: 301-587-1788 (Purple/ZVRS); 301-328-1443 (Sorenson); 301-338-6380 (Convo)Fax: 301-587-1791Email: nad.info@nad.org
- National Federation of the BlindPhone: 410-659-9314Email: nfb@nfb.org
- Unique: Understanding Rare Chromosome and Gene DisordersUnited KingdomPhone: +44 (0) 1883 723356Email: info@rarechromo.org
- Norrie Disease RegistryMassachusetts General Hospital185 Cambridge StreetCRP Building North, 5th Floor, Suite 5300Boston MA 02114Phone: 617-726-5718Fax: 617-724-9620Email: ksims@mgh.harvard.edu
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
NDP-Related Retinopathies: Genes and Databases
Table B.
OMIM Entries for NDP-Related Retinopathies (View All in OMIM)
Molecular Pathogenesis
NDP encodes norrin, a secreted protein with a cysteine-knot motif (highly conserved in many growth factors), which is expressed widely in brain astrocytes and cerebellar Bergmann glia [Ye et al 2011]. Norrin is a high-affinity ligand for frizzled-4, resulting in activation of the canonic Wnt pathway. The Wnt pathway regulates retinal angiogenesis. NDP loss-of-function variants impair retinal angiogenesis by either abolishing gene function or altering frizzled-4 binding [Xu et al 2004, Rattner et al 2014].
NDP pathogenic missense variants often affect one of the many cysteine residues immediately adjacent to a cysteine. These cysteine residues are presumed important for the maintenance of protein structure. The norrin protein has a highly conserved cysteine-knot motif (exon 3). The cysteine knot (active domain) provides the structural conformation required for receptor binding for activation of the Wnt receptor–𝛽-catenin signal transduction pathway [Wu et al 2007].
Norrin is critical in central nervous system vascular development, as it is required for:
- Angiogenesis in the eye, ear, and brain;
- Maintenance of the blood-brain barrier and the blood-retinal barrier;
- Neuro-protective properties for retinal neurons.
It has been shown that norrin protects against oxidative damage of retinal cells, and pathogenic NDP variants worsen the severity of retinopathy of prematurity in premature infants.
Audiologic and animal model data suggest that the pathology resides in the cochlea (specifically, the stria vascularis) and that retrocochlear and brain auditory system function is normal [Rehm et al 2002, Bryant et al 2022]. A recent study demonstrated that norrin controls a network of transcriptional regulators required for maturation and maintenance of cochlear hair cells, and overexpression of Ndp rescued hair cells and cochlear function in Ndp knockout mice [Hayashi et al 2021].
The role of norrin in intellectual disability is unknown [McNeill et al 2013].
Mechanism of disease causation. Loss of function
Table 7.
NDP-Related Retinopathies: Gene-Specific Laboratory Considerations
Chapter Notes
Author History
Lisa A Schimmenti, MD (2022-present)
Brittni A Scruggs, MD, PhD (2022-present)
Madeline Q Reding, MS, MPH, CGC (2022-present)
Katherine B Sims, MD; Massachusetts General Hospital (1999-2022)
Revision History
- 23 March 2023 (aa/gm) Revision: Daich Varela et al [2023] and information about likely pathogenic variants in noncoding regions detected by genome sequencing added to Establishing the Diagnosis and Molecular Genetics
- 31 March 2022 (bp) Comprehensive update posted live
- 18 September 2014 (me) Comprehensive update posted live
- 23 July 2009 (me) Comprehensive update posted live
- 8 August 2006 (me) Comprehensive update posted live
- 14 May 2004 (me) Comprehensive update posted live
- 11 June 2002 (me) Comprehensive update posted live
- 30 July 1999 (me) Review posted live
- 10 February 1999 (ks) Original submission
References
Literature Cited
- Allen RC, Russell SR, Streb LM, Alsheikheh A, Stone EM. Phenotypic heterogeneity associated with a novel mutation (Gly112Glu) in the Norrie disease protein. Eye. 2006;20:234–41. [PubMed: 15776010]
- Arai E, Fujimaki T, Yanagawa A, Fujiki K, Yokoyama T, Okumura A, Shimizu T, Murakami A. Familial cases of Norrie disease detected by copy number analysis. Jpn J Ophthalmol. 2014;58:448–54. [PubMed: 25023092]
- Black GC, Perveen R, Bonshek R, Cahill M, Clayton-Smith J, Lloyd IC, McLeod D. Coats' disease of the retina (unilateral retinal telangiectasis) caused by somatic mutation in the NDP gene: a role for norrin in retinal angiogenesis. Hum Mol Genet. 1999;8:2031–5. [PubMed: 10484772]
- Bortolato M, Floris G, Shih JC. From aggression to autism: new perspectives on the behavioral sequelae of monoamine oxidase deficiency. J Neural Transm (Vienna). 2018;125:1589–99. [PMC free article: PMC6215718] [PubMed: 29748850]
- Bryant D, Pauzuolyte V, Ingham NJ, Patel A, Pagarkar W, Anderson LA, Smith KE, Moulding DA, Leong YC, Jafree DJ, Long DA, Al-Yassin A, Steel KP, Jagger DJ, Forge A, Berger W, Sowden JC, Bitner-Glindzicz M. The timing of auditory sensory deficits in Norrie disease has implications for therapeutic intervention. JCI Insight. 2022;7:e148586. [PMC free article: PMC8855802] [PubMed: 35132964]
- Chow CC, Kiernan DF, Chau FY, Blair MP, Ticho BH, Galasso JM, Sahpiro MJ. Laser photocoagulation at birth prevents blindness in Norrie's disease diagnosed using amniocentesis. Ophthalmology. 2010;117:2402–6. [PubMed: 20619898]
- Coussa RG, Zhao Y, DeBenedictis MJ, Babiuch A, Sears J, Traboulsi EI. Novel mutation in CTNNB1 causes familial exudative vitreoretinopathy (FEVR) and microcephaly: case report and review of the literature. Ophthalmic Genet. 2020;41:63–8. [PubMed: 32039639]
- Daich Varela M, Bellingham J, Motta F, Jurkute N, Ellingford JM, Quinodoz M, Oprych K, Niblock M, Janeschitz-Kriegl L, Kaminska K, Cancellieri F, Scholl HPN, Lenassi E, Schiff E, Knight H, Black G, Rivolta C, Cheetham ME, Michaelides M, Mahroo OA, Moore AT, Webster AR, Arno G. Multidisciplinary team directed analysis of whole genome sequencing reveals pathogenic non-coding variants in molecularly undiagnosed inherited retinal dystrophies. Hum Mol Genet. 2023;32:595–607. [PMC free article: PMC9896476] [PubMed: 36084042]
- Davitt BV, Morgan C, Cruz OA. Sleep disorders in children with congenital anophthalmia and microphthalmia. J AAPOS. 1997;1:151–3. [PubMed: 10532777]
- Dickinson JL, Sale MM, Passmore A, Fitzgerald LM, Wheatley CM, Burdon KP, Craig JE, Tengtrisorn S, Carden SM, Maclean H, Mackey DA. Mutations in the NDP gene: contribution to Norrie disease, familial exudative vitreoretinopathy and retinopathy of prematurity. Clin Exp Ophthalmol. 2006;34:682–8. [PubMed: 16970763]
- Dixon MW, Stem MS, Schuette JL, Keegan CE, Besirli CG. CTNNB1 mutation associated with familial exudative vitreoretinopathy (FEVR) phenotype. Ophthalmic Genet. 2016;37:468–70. [PubMed: 26967979]
- Drenser KA, Fecko A, Dailey W, Trese MT. A characteristic phenotypic retinal appearance in Norrie disease. Retina. 2007;27:243–6. [PubMed: 17290208]
- Dubucs C, Merveille M, Kessler S, Sevely A, Chassaing N, Calvas P. Prenatal diagnosis of Norrie disease based on ultrasound findings. Ultrasound Obstet Gynecol. 2019;54:138–9. [PubMed: 30125416]
- Ghosh M, Sharma S, Shastri S, Arora S, Shukla R, Gupta N, Deka D, Kabra M. Norrie disease: first mutation report and prenatal diagnosis in an Indian family. Indian J Pediatr. 2012;79:1529–31. [PubMed: 22674248]
- Halpin C, Owen G, Gutierrez-Espeleta GA, Sims K, Rehm HL. Audiologic features of Norrie disease. Ann Otol Rhinol Laryngol. 2005;114:533–8. [PubMed: 16134349]
- Halpin C, Sims K. Twenty years of audiology in a patient with Norrie disease. Int J Pediatr Otorhinolaryngol. 2008;72:1705–10. [PubMed: 18817988]
- Han S, Sun J, Yang L, Qi M. Role of NDP- and FZD4-related novel mutations identified in patients with FEVR in norrin/β-catenin signaling pathway. Biomed Res Int. 2020;2020:7681926. [PMC free article: PMC7201721] [PubMed: 32420371]
- Hayashi Y, Chiang H, Tian C, Indzhykulian A, Edge A. Norrie disease protein is essential for cochlear hair cell maturation. Proc Natl Acad Sci U S A. 2021;118:e2106369118. [PMC free article: PMC8488680] [PubMed: 34544869]
- Hiraoka M, Berinstein DM, Trese MT, Shastry BS. Insertion and deletion mutations in the dinucleotide repeat region of the Norrie disease gene in patients with advanced retinopathy of prematurity. J Hum Genet. 2001;46:178–81. [PubMed: 11322656]
- Hiraoka M, Takahashi H, Orimo H, Hiraoka M, Ogata T, Azuma N. Genetic screening of Wnt signaling factors in advanced retinopathy of prematurity. Mol Vis. 2010;16:2572–7. [PMC free article: PMC3000231] [PubMed: 21151595]
- Jia B, Huang L, Chen Y, Liu S, Chen C, Xiong K, Song L, Zhou Y, Yang X, Zhong M. A novel contiguous deletion involving NDP, MAOB and EFHC2 gene in a patient with familial Norrie disease: bilateral blindness and leucocoria without other deficits. J Genet. 2017;96:1015–20. [PubMed: 29321361]
- Khan AO, Shamsi FA, Al-Saif A, Kambouris M. A novel missense Norrie disease mutation associated with a severe ocular phenotype. J Pediatr Ophthalmol Strabismus. 2004;41:361–3. [PubMed: 15609522]
- Li Y, Li J, Zhang X, Peng J, Li J, Zhao P. Identification of gene mutations in atypical retinopathy of prematurity cases. J Ophthalmol. 2020;2020:4212158. [PMC free article: PMC7455826] [PubMed: 32884843]
- Lin P, Shankar SP, Duncan J, Slavotinek A, Stone EM, Rutar T. Retinal vascular abnormalities and dragged maculae in a carrier with a new NDP mutation (c.268delC) that caused severe Norrie disease in the proband. J AAPOS. 2010;14:93–6. [PubMed: 20227630]
- McNeill B, Mazerolle C, Bassett EA, Mears AJ, Ringuette R, Lagali P, Picketts DJ, Paes K, Rice D, Wallace VA. Nedgehog regulates Norrie disease protein to drive neural progenitor self-renewal. Hum Mol Genet. 2013;22:1005–16. [PubMed: 23201751]
- Meire FM, Lafaut BA, Speleman F, Hanssens M. Isolated Norrie disease in a female caused by a balanced translocation t(X,6). Ophthalmic Genet. 1998;19:203–7. [PubMed: 9895245]
- Michaelides M, Luthert PJ, Cooling R, Firth H, Moore AT. Norrie disease and peripheral venous insufficiency. Br J Ophthalmol. 2004;88:1475. [PMC free article: PMC1772398] [PubMed: 15489496]
- Nikopoulos K, Venselaar H, Collin RW, Riveiro-Alvarez R, Boonstra FN, Hooymans JM, Mukhopadhyay A, Shears D, van Bers M, de Wijs IJ, van Essen AJ, Sijmons RH, Tilanus MA, van Nouhuys CE, Ayuso C, Hoefsloot LH, Cremers FP. Overview of the mutation spectrum in familial exudative vitreoretinopathy and Norrie disease with identification of 21 novel variants in FZD4, LRP5, and NDP. Hum Mutat. 2010;31:656–66. [PubMed: 20340138]
- Parzefall T, Lucas T, Ritter M, Ludwig M, Ramsebner R, Frohne A, Schofer C, Hengstschlager M, Frei K. A novel missense NDP mutation [p.(Cys93Arg)] with a manifesting carrier in an Austrian family with Norrie disease. Audiol Neurootol. 2014;19:203–9. [PubMed: 24801666]
- Rattner A, Wang Y, Zhou Y, Williams J, Nathans J. The role of the hypoxia response in shaping retinal vascular development in the absence of Norrin/Frizzled4 signaling. Invest Ophthalmol Vis Sci. 2014;55:8614–25. [PMC free article: PMC4280883] [PubMed: 25414188]
- Redmond RM, Vaughan JI, Jay M, Jay B. In-utero diagnosis of Norrie disease by ultrasonography. Ophthalmic Paediatr Genet. 1993;14:1–3. [PubMed: 8345950]
- Rehm HL, Zhang DS, Brown MC, Burgess B, Halpin C, Berger W, Morton CC, Corey DP, Chen ZY. Vascular defects and sensorineural deafness in a mouse model of Norrie disease. J Neurosci. 2002;22:4286–92. [PMC free article: PMC6758776] [PubMed: 12040033]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rodriguez-Revenga L, Madrigal I, Alkhalidi LS, Armengol L, Gonzalex E, Badenas C, Estivill X, Mila M. Contiguous deletion of the NDP, MAOA, MAOBV and EFHC2 genes in a patient with Norrie disease, severe psychomotor retardation and myoclonic epilepsy. Am J Med Genet A. 2007;143A:916–20. [PubMed: 17431911]
- Royer G, Hanein S, Raclin V, Gigarel N, Rozet JM, Munnich A, Steffann J, Dufier JL, Kaplan J, Bonnefont JP. NDP gene mutations in 14 French families with Norrie disease. Hum Mutat. 2003;22:499. [PubMed: 14635119]
- Schuback DE, Chen ZY, Craig IW, Breakefield XO, Sims KB. Mutations in the Norrie disease gene. Hum Mutat. 1995;5:285–92. [PubMed: 7627181]
- Shastry BS, Hiraoka M, Trese DC, Trese MT. Norrie disease and exudative vitreoretinopathy in families with affected female carriers. Eur J Ophthalmol. 1999;9:238–42. [PubMed: 10544980]
- Shastry BS. Persistent hyperplastic primary vitreous: congenital malformation of the eye. Clin Experiment Ophthalmol. 2009;37:884–90. [PubMed: 20092598]
- Shima C, Kusaka S, Kondo H, Hasebe H, Fujikado T, Tano Y. Lens-sparing vitrectomy effective for reattachment of newly developed falciform retinal detachment in a patient with Norrie disease. Arch Ophthalmol. 2009;127:579–80. [PubMed: 19365046]
- Shute CL, McLoone E. Reaching a FEVR pitch: a case series of familial exudative vitreoretinopathy in Northern Ireland. J Pediatr Ophthalmol Strabismus. 2022;59:102–9. [PubMed: 34592872]
- Sims KB, de la Chapelle A, Norio R, Sankila EM, Hsu YP, Rinehart WB, Corey TJ, Ozelius L, Powell JF, Bruns G, Gusella JF, Murphy DL, Breakefield XO. Monoamine oxidase deficiency in males with an X chromosome deletion. Neuron. 1989;2:1069–76. [PubMed: 2483108]
- Sisk RA, Hufnagel RB, Bandi S, Polzin WJ, Ahmed ZM. Planned Preterm Delivery and Treament of retinal neovascularization in Norrie disease. Ophthalmology. 2014;121:1312–3. [PMC free article: PMC9126609] [PubMed: 24529712]
- Smith SE, Mullen TE, Graham D, Sims KB, Rehm HL. Norrie disease: extraocular clinical manifestations in 56 patients. Am J Med Genet. 2012;158A:1909–17. [PubMed: 22786811]
- Sowden JC, Kros CJ, Sirimanna T, Pagarkar W, Oluonye N, Henderson RH. Impact of sight and hearing loss in patients with Norrie disease: advantages of dual sensory clinics in patient care. BMJ Paediatr Open. 2020;4:e000781. [PMC free article: PMC7670942] [PubMed: 33225082]
- Suárez-Merino B, Bye J, McDowall J, Ross M, Craig IW. Sequence analysis and transcript identification within 1.5 mB of DNA deleted together with the NDP and MAO genes in atypical Norrie disease patients presenting with a profound phenotype. Hum Mutat. 2001;17:523. [PubMed: 11385715]
- Sudha D, Ganapathy A, Mohan P, Mannan AU, Krishna S, Neriyanuri S, Swaminathan M, Rishi P, Chidambaram S, Arunachalam JP. Clinical and genetic analysis of Indian patients with NDP-related retinopathies. Int Ophthalmol. 2018;38:1251–60. [PubMed: 28602015]
- Talebi F, Ghanbari Mardasi F, Mohammadi Asl J, Lashgari A, Farhadi F. Identification of a novel missense mutation in the Norrie disease gene: the first molecular genetic analysis and prenatal diagnosis of Norrie disease in an Iranian family. Cell J. 2018;20:290–2. [PMC free article: PMC5893302] [PubMed: 29633608]
- Taylor JC, Martin HC, Lise S, Broxholme J, Cazier JB, Rimmer A, Kanapin A, Lunter G, Fiddy S, Allan C, Aricescu AR, Attar M, Babbs C, Becq J, Beeson D, Bento C, Bignell P, Blair E, Buckle VJ, Bull K, Cais O, Cario H, Chapel H, Copley RR, Cornall R, Craft J, Dahan K, Davenport EE, Dendrou C, Devuyst O, Fenwick AL, Flint J, Fugger L, Gilbert RD, Goriely A, Green A, Greger IH, Grocock R, Gruszczyk AV, Hastings R, Hatton E, Higgs D, Hill A, Holmes C, Howard M, Hughes L, Humburg P, Johnson D, Karpe F, Kingsbury Z, Kini U, Knight JC, Krohn J, Lamble S, Langman C, Lonie L, Luck J, McCarthy D, McGowan SJ, McMullin MF, Miller KA, Murray L, Németh AH, Nesbit MA, Nutt D, Ormondroyd E, Oturai AB, Pagnamenta A, Patel SY, Percy M, Petousi N, Piazza P, Piret SE, Polanco-Echeverry G, Popitsch N, Powrie F, Pugh C, Quek L, Robbins PA, Robson K, Russo A, Sahgal N, van Schouwenburg PA, Schuh A, Silverman E, Simmons A, Sørensen PS, Sweeney E, Taylor J, Thakker RV, Tomlinson I, Trebes A, Twigg SR, Uhlig HH, Vyas P, Vyse T, Wall SA, Watkins H, Whyte MP, Witty L, Wright B, Yau C, Buck D, Humphray S, Ratcliffe PJ, Bell JI, Wilkie AO, Bentley D, Donnelly P, McVean G. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet. 2015;47:717–26. [PMC free article: PMC4601524] [PubMed: 25985138]
- Walsh MK, Drenser KA, Capone A Jr, Trese MT. Early vitrectomy effective for Norrie disease. Arch Ophthalmol. 2010;128:456–60. [PubMed: 20385941]
- Wu LH, Chen LH, Xie H, Xie YJ. Prenatal diagnosis of a case of Norrie disease with late development of bilateral ocular malformation. Fetal Pediatr Pathol. 2017;36:240–5. [PubMed: 28394646]
- Wu W-C, Drenser K, Trese M, Capone A Jr, Dailey W. Retinal phenotype-genotype correlation of pediatric patients expressing mutations in the Norrie disease gene. Arch Ophthalmol. 2007;125:225–30. [PubMed: 17296899]
- Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K, Nathans J. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–95. [PubMed: 15035989]
- Yamada K, Limprasert P, Ratanasukon M, Tengtrisorn S, Yingchareonpukdee J, Vasiknanonte P, Kitaoka T, Ghadami M, Niikawa N, Kishino T. Two Thai families with Norrie disease (ND): association of two novel missense mutations with severe ND phenotype, seizures, and a manifesting carrier. Am J Med Genet. 2001;100:52–5. [PubMed: 11337749]
- Yang H, Li S, Xiao X, Guo X, Zhange Q. Screening for DNP mutations in 44 unrelated pateints with familial exudative vitreoretinopathy or Norrie disease. Curr Eye Res. 2012;37:726–9. [PubMed: 22563645]
- Yang J, Xiao X, Li S, Mai G, Jia X, Wang P, Sun W, Zhang Q. Severe exudative vitreoretinopathy as a common feature for CTNNB1, KIF11 and NDP variants plus sector degeneration for KIF11. Am J Ophthalmol. 2022;235:178–87. [PubMed: 34582765]
- Ye X, Smallwood P, Nathans J. Expression of the Norrie disease gene (Ndp) in developing and adult mouse eye, ear, and brain. Gene Expr Patterns. 2011;11:151–5. [PMC free article: PMC3061303] [PubMed: 21055480]
- Yonekawa Y, Wu WC, Kusaka S, Robinson J, Tsujioka D, Kang KB, Shapiro MJ, Padhi TR, Jain L, Sears JE, Kuriyan AE, Berrocal AM, Quiram PA, Gerber AE, Paul Chan RV, Jonas KE, Wong SC, Patel CK, Abbey AM, Spencer R, Blair MP, Chang EY, Papakostas TD, Vavvas DG, Sisk RA, Ferrone PJ, Henderson RH, Olsen KR, Hartnett ME, Chau FY, Mukai S, Murray TG, Thomas BJ, Meza PA, Drenser KA, Trese MT, Capone A Jr. Immediate sequential bilateral pediatric vitreoretinal surgery: an international multicenter study. Ophthalmology. 2016;123:1802–8. [PMC free article: PMC5522749] [PubMed: 27221737]
- Zhang XY, Jiang WY, Chen LM, Chen SQ. A novel Norrie disease pseudoglioma gene mutation, c.-1_2delAAT, responsible for Norrie disease in a Chinese family. Int J Ophthalmol. 2013;6:739–43. [PMC free article: PMC3874509] [PubMed: 24392318]
Publication Details
Author Information and Affiliations
Vitreoretinal Surgery & Diseases
Department of Ophthalmology
Mayo Clinic
Rochester, Minnesota
Assistant Dean, MD/PhD Affairs MCGSBS
Professor, Pediatrics Consultant, Departments of Clinical Genomics, Otorhinolaryngology, Head and Neck Surgery, Ophthalmology, and Biochemistry and Molecular Biology
Mayo Clinic School of Medicine
Rochester, Minnesota
Publication History
Initial Posting: July 30, 1999; Last Revision: March 23, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Scruggs BA, Reding MQ, Schimmenti LA. NDP-Related Retinopathies. 1999 Jul 30 [Updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

