Summary
Clinical description.
Primary hyperoxaluria type 3 (PH3) is characterized by recurring calcium oxalate stones beginning in childhood or adolescence and, on occasion, nephrocalcinosis or reduced kidney function. PH3 most often presents in childhood (median age 2 to 3 years) with signs or symptoms related to stones including hematuria, frequent urination, dysuria, blood visible in the urine, or stone-associated pain. Some individuals with PH3 do not present until adulthood, usually with stone-related symptoms or findings. Over time, frequent stones and/or nephrocalcinosis may compromise kidney function, resulting in chronic kidney disease. To date, systemic oxalosis has not been reported in PH3.
Management.
Treatment of manifestations: There is no cure for PH3. Lifelong treatment includes medical therapy and pharmacotherapy to reduce urine supersaturation of calcium oxalate to prevent formation of stones and calcium oxalate crystals that can injure the kidney. Mainstays of treatment are high oral fluid intake (>2.5 L per m2 body surface area) at all times; oral administration of an inhibitor of calcium oxalate crystallization, typically potassium and/or sodium citrate; prevention of stone complications by prompt relief of urinary tract obstruction and treatment of urinary tract infections.
Surveillance: For those who are stable: (1) annual clinical assessment of stone-related symptoms (pain); frequency of passage of urinary stones and/or gravel and urinary tract infection; adherence to high fluid intake and medication schedule; and (2) annual assessment of kidney function (serum creatinine and eGFR), measurement of plasma oxalate concentration in those with impaired renal function, 24-hour urine oxalate and supersaturation study, and renal ultrasound examination or other imaging to monitor for stone formation.
More frequent assessments are required for: children under age four years, individuals with complex stone problems, and individuals with reduced kidney function.
Agents/circumstances to avoid: Intravascular volume contraction, delays in treatment of acute stone episodes, nephrotoxic agents, high-dose ascorbic acid, and marked dietary oxalate excess.
Evaluation of relatives at risk: Targeted molecular genetic testing for the familial HOGA1 pathogenic variants is recommended for all sibs of a proband (regardless of age and even if apparently asymptomatic) in order to identify as early as possible those who would benefit from early treatment, preventive measures, and knowledge of circumstances/agents to avoid.
Pregnancy management: In the few reports available to date, pregnancy outcomes in women with PH3 appear to be similar to those in women with other genetic causes of hyperoxaluria. Nonetheless, pregnant women who have PH3 should be considered at higher risk and warrant close monitoring, given the increased risk of acute kidney injury due to hypovolemia and/or an obstructing or infected stone.
Genetic counseling.
PH3 is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a HOGA1 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the HOGA1 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal/preimplantation genetic testing are possible.
Diagnosis
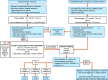
For recently published algorithms for the diagnosis of the primary hyperoxalurias (PH), see [Singh et al 2022a].
Algorithm for the diagnostic evaluation of primary hyperoxaluria in an affected individual * Random oxalate-to-creatinine ratios vary significantly by age. Consult pediatric reference range tables for interpretation.
Suggestive Findings
Primary hyperoxaluria type 3 (PH3) should be considered in a proband with the following clinical, radiographic, and/or laboratory findings and family history.
Clinical and radiographic findings
Calcium oxalate stones, especially when in both kidneys
Recurring calcium oxalate stones
Onset of stone disease in childhood or adolescence
Reduced kidney function in the presence of calcium stones or nephrocalcinosis (crystal deposition in renal parenchyma)
Nephrocalcinosis
Laboratory findings
Urine oxalate. >0.7 mmol per 1.73 m
2 per 24 hours in individuals with preserved kidney function (glomerular filtration rate [GFR] >40 mL/min per 1.7 m
2). Note: Hyperoxaluria, which tends to be more variable in PH3 than in
PH1 and
PH2, may intermittently be in the range of 0.4-0.7 mmol per 1.73 m
2 per 24 hrs.
Note: (1) Normal urine oxalate is <0.46 mmol per 1.73 m
2 per 24 hours. Values higher than the normal range should be repeated to detect persistent hyperoxaluria that may intermittently increase to a level indicative of PH. (2) Urine oxalate may be lower in individuals with advanced chronic kidney disease (CKD). (3) In children, the oxalate excretion rate must be corrected for 1.73 m
2 body surface area. (4) The individual should not be receiving pyridoxine or vitamin supplements when urine and plasma oxalate levels are used for the purpose of diagnosis. (5) Urine oxalate should preferably be measured in a 24-hour urine sample; however, when timed urine collections cannot be obtained, a random urine oxalate-to-creatinine ratio can be used. (6) Since normal ranges for the oxalate-to-creatinine ratio vary during childhood by age, age-related normal values should be consulted for accurate interpretation (see
Table 1).
Plasma oxalate. Mild elevation of plasma oxalate (1.6-20 µmol/L) is often seen in individuals with PH3 with CKD stages 1-3A [
Martin-Higueras et al 2021,
Singh et al 2022a]. Plasma oxalate may be higher in individuals with advanced CKD (GFR <45 mL/min per 1.73 m
2).
Note: (1) Normal plasma oxalate concentration varies depending on methods of sample preparation and measurement [
Stokes et al 2020]. (2) The individual being tested should not be receiving pyridoxine, high doses of ascorbic acid, or other vitamin supplements when urine and plasma oxalate are obtained for the purpose of diagnosis.
Urine 4-hydroxyoxoglutarate (HOG) or dihydroxyglutarate (DHG), measured in a random (spot) urine sample, are increased in most individuals with PH3 and appear to be sensitive diagnostic markers [
Ventzke et al 2017,
Greed et al 2018,
Woodward et al 2019]. Urine HOG-to-creatinine and DHG-to-creatinine ratios vary by age. Laboratory-specific normal values by age should be consulted.
Table 1.
Random Urine Oxalate-to-Creatinine Ratio in Children by Age
View in own window
| Age | Upper Limit of Normal 2 |
|---|
| (mmol/mmol) | (mg/mg) |
|---|
| <6 months 1 | 0.37 | 0.29 |
| 6 months to 2 years | 0.26 | 0.20 |
| >2 years to 5 years | 0.14 | 0.11 |
| 6 to 12 years | 0.08 | 0.06 |
- 1.
Urine oxalate-to-creatinine ratios are higher in very premature infants than in term infants, especially when they are receiving parenteral nutrition containing amino acids. The ratio falls when premature infants are receiving only glucose and electrolyte solutions [Campfield & Braden 1989].
- 2.
When very high dietary intake of oxalate or low dietary intake of calcium is suspected as the cause of the hyperoxaluria, the diet should be corrected and the urine oxalate remeasured for verification.
Family history. Family history of recurring calcium oxalate stones and/or chronic kidney disease that is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). HOGA1 founder variants have been identified in individuals of Ashkenazi Jewish heritage (see Table 4). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of PH3 is established in a proband with suggestive findings and biallelic pathogenic (or likely pathogenic) variants in HOGA1 identified by molecular genetic testing (see Table 2).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic HOGA1 variants of uncertain significance (or of one known HOGA1 pathogenic variant and one HOGA1 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (targeted analysis for founder variants [see Table 4], multigene panel) and comprehensive
genomic testing (exome sequencing, genome sequencing).
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1) whereas genomic testing does not (see Option 2).
Option 1
A nephrology or monogenic urinary stone
multigene panel that includes HOGA1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive
genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 2.
Molecular Genetic Testing Used in Primary Hyperoxaluria Type 3
View in own window
| Gene 1 | Method | Proportion of Pathogenic Variants 2 Detectable by Method |
|---|
|
HOGA1
| Sequence analysis 3 | All pathogenic variants reported to date 4 |
| Gene-targeted deletion/duplication analysis 5 | None reported to date 4 |
- 1.
- 2.
- 3.
- 4.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and gene-targeted microarray designed to detect single-exon deletions or duplications.
Clinical Characteristics
Clinical Description
Primary hyperoxaluria type 3 (PH3) is characterized by recurring calcium oxalate kidney stones beginning in childhood or adolescence and, on occasion, nephrocalcinosis or reduced kidney function. In individuals with PH3, stone formation typically begins prior to age five years, though in some individuals stones may not be clinically evident until adulthood [Martin-Higueras et al 2021, Singh et al 2022b, Arnous et al 2023].
PH3 most often presents in childhood (median age 2 to 3 years) with signs or symptoms related to stones including hematuria, frequent urination, dysuria, blood visible in the urine, or stone-associated pain. In one large study the median number of stones present at first imaging in individuals with PH3 was four [Arnous et al 2023].
The lifelong clinical stone burden is substantial, as symptomatic stone events recur through the sixth decade of life [Martin-Higueras et al 2021, Singh et al 2022b], many of which require surgery for stone removal.
Some individuals with PH3 do not present until adulthood, usually with stone-related symptoms or findings. Although stones are likely to be discovered due to symptoms, they may also be detected incidentally on imaging studies performed for other purposes.
Over time, frequent stones and/or nephrocalcinosis may compromise kidney function, resulting in chronic kidney disease (CKD). Individuals with PH3 who are older than age 40 years may have CKD stage 3 or higher that exceeds the expected age-related glomerular filtration rate (GFR) decline [Singh et al 2022b]. Nephrocalcinosis, reported in 7%-10% of individuals with PH3, resulted in an estimated GFR (eGFR) that was lower in individuals with nephrocalcinosis than in those without it [Singh et al 2022b]. Nonetheless, Singh et al [2022b] reported that just 3% of individuals with PH3 had progressed to kidney failure by age 40 years, compared with 64% of individuals with PH1 and 34% with PH2.
To date, the only three individuals with PH3 reported to progress to kidney failure also had other factors that may have contributed to CKD progression. These individuals were:
An eight-year-old who had multiple stone removal procedures and urinary tract obstruction [
Hopp et al 2015];
A 78-year-old male who had a 30-year history of nephrolithiasis who developed end-stage kidney disease after unilateral nephrectomy for clear cell carcinoma [
Richard et al 2017].
Systemic oxalosis has not been reported, though the number of individuals with PH3 with advanced CKD reported to date is small.
Heterozygotes. Although some heterozygotes (carriers) demonstrate elevated urinary precursors of oxalate [Pitt et al 2015], they do not appear to have hyperoxaluria or a stone event rate above that expected in the general population [Bar et al 2021].
Prevalence
Surveys of clinicians in central Europe led to estimates of the prevalence of primary hyperoxaluria (PH) of all causes of one to three in 1,000,000 [Cochat & Rumsby 2013].
Among individuals with primary hyperoxaluria, approximately 70% have PH1, 10% have PH2, 7%-12% have PH3, and 10% have an unidentified genetic cause [Cochat & Rumsby 2013, Hopp et al 2015, Martin-Higueras et al 2021, Singh et al 2022b].
Registry data from the Rare Kidney Stone Consortium and OxalEurope suggest that the prevalence of PH3 is similar to that of PH2 and approximately one sixth that of PH1.
By contrast, estimates from publicly available population data (NHLBI Exome Sequencing Project) showed an estimated prevalence of PH3 (based on genomic data) of one in 136,000 [Hopp et al 2015].
The PH3 carrier frequency is one in 185 [Hopp et al 2015], which is similar to the rate observed for PH1, suggesting that individuals with PH3 either remain undiagnosed or do not have clinical manifestations.
PH3 has been observed more commonly among individuals of Ashkenazi Jewish descent, and a 3-bp deletion founder variant has been identified [Belostotsky et al 2010]. Although pathogenic HOGA1 variants have a carrier frequency of one in 55 in Israeli Ashkenazi Jews [Bar et al 2021], the true prevalence in this population is unknown (see Table 4).
Differential Diagnosis
Primary Hyperoxalurias
Primary hyperoxaluria (PH) should be included in the differential diagnosis of any condition that causes calcium oxalate kidney stone disease or nephrocalcinosis and is associated with hyperoxaluria. The three known types of PH are PH1 (due to biallelic AGXT pathogenic variants), PH2 (due to biallelic GRHPR pathogenic variants), and PH3 (due to biallelic HOGA1 pathogenic variants). Each gene encodes an enzyme for different metabolic pathways relevant for the metabolism of glyoxylate [Cochat & Rumsby 2013]. Although oxalate is believed to accumulate as an end product in all forms of PH, in PH3 the biochemical mechanism whereby deficiency of 4-hydroxy-2-oxoglutarate aldolase (HOGA) results in increased oxalate production is unclear [Dindo et al 2019, Bar et al 2021].
Of the primary hyperoxalurias, PH1 accounts for approximately 70%, PH2 for 10%, PH3 for 10%, and 10% do not have an identified genetic cause to date [Hopp et al 2015]. The clinical manifestations of the three known types of PH overlap considerably (see Table 3). One recent series found urine oxalate to be lower in PH3 than PH1 and PH2 in a predominantly North American PH population [Singh et al 2022a], whereas a largely European case series observed similar oxalate excretion rates among all three PH types [Martin-Higueras et al 2021]. In both series urine citrate and calcium excretion were normal in individuals with PH3. These observations in individuals with PH3 contrast with individuals with PH1, who often manifest lower urine citrate and calcium excretion rates [Singh et al 2022b].
Although increased urinary excretion of the following specific organic acids are suggestive of PH type [Woodward et al 2019], definitive diagnosis requires genetic testing:
Table 3.
Comparison of Primary Hyperoxaluria Types 1, 2, and 3
View in own window
Gene
(Disorder) | Age of 1st Symptoms 1 | Nephrocalcinosis | eGFR 1 at Diagnosis
(mL/min per 1.73 m2) | Plasma Oxalate 1
(µmol/L; nL <1.6) | Urine 1 | ESKD
at Age 40 Yrs |
|---|
Oxalate
(mmol per 1.73 m2 per 24 hrs; nL <0.46) | Calcium
(mg per 1.73 m2 per 24 hrs; nL = 100-300) | Citrate
(mg per 1.73 m2 per 24 hrs; nL = 320-1240) |
|---|
AGXT
(PH1) | Age 4.9 yrs | 25.5% | 48 | 12.5 | 1.6 | 51 | 255 | ~64% |
GRHPR
(PH2) | Age 5.7 yrs | 15.7% | 83 | 4.3 | 1.5 | 98 | 717 | ~34% |
HOGA1
(PH3) | Age 2.7 yrs | 6.5% | 96 | 2.1 | 1.1 | 112 | 638 | ~3% |
eGFR = estimated glomerular filtration rate; ESKD = end-stage kidney disease; nL = normal limit
- 1.
Parameters presented are the median.
Idiopathic calcium oxalate stone disease can be associated with mild hyperoxaluria. In individuals who form idiopathic stones, hyperoxaluria (1) is typically less (<0.6 mmol/day) than that observed in individuals with a primary hyperoxaluria; (2) varies from one collection to the next; and (3) is frequently associated with mild hypercalciuria. Since idiopathic stone disease is very common and since calcium oxalate is found in approximately 80% of stones [Worcester & Coe 2010], a high index of clinical suspicion is necessary to identify the small proportion of individuals who form calcium oxalate stones as a result of PH3.
Secondary Hyperoxalurias
Secondary forms of hyperoxaluria should be systematically considered in the differential diagnosis [Lumlertgul et al 2018].
Dietary or other sources of excessive oxalate or oxalate precursors include:
Foods high in oxalate, especially if dietary calcium intake is low;
Marked deficiency of dietary calcium, which leaves a greater proportion of oxalate free in the intestinal lumen, resulting in increased absorption of oxalate and hyperoxaluria;
Very high doses of vitamin C;
Toxins, such as ethylene glycol, which can cause marked hyperoxaluria and associated acute renal failure.
Enteric hyperoxaluria results from fat malabsorption in the small intestine. Undigested fat reaching the colon combines with calcium, thus decreasing the amount of calcium available to bind to oxalate, which is subsequently absorbed. Fatty acids not absorbed in the small intestine can damage the colonic mucosa, leading to further increase in oxalate absorption. Causes include the following:
Other Monogenic Stone Diseases
Phenotypic overlap with other monogenic stone diseases can occur [Cogal et al 2021], particularly those characterized by childhood onset of calcium-containing stones and nephrocalcinosis. They can be differentiated from PH3 by the absence of hyperoxaluria. Nephrology or monogenic stone multigene genetic testing panels are particularly valuable for definitive diagnosis in this setting.
Nephrocalcinosis of Prematurity
Nephrocalcinosis of prematurity, which occurs in a significant proportion of infants born prior to 28 weeks' gestation, is also characterized by nephrolithiasis [Habbig et al 2011]. Contributing risk factors in premature infants are thought to include: (1) urine oxalate that is higher than that observed in infants born at term [Schell-Feith et al 2010]; (2) hypercalciuria and hypocitric aciduria. Since individuals with PH3 often develop stones in infancy or during early childhood [Milliner & Matsumoto 2020, Singh et al 2022b], there may be confusion with stones or nephrocalcinosis related to prematurity.
Management
No clinical practice guidelines specific to primary hyperoxaluria type 3 (PH3) have been published, though general recommendations for individuals with all forms of PH are relevant [Groothoff et al 2023]. The following recommendations are also based on the authors' experience and participation in the Rare Kidney Stone Consortium PH Registry, the Oxalosis and Hyperoxaluria Foundation working group on practice guidelines, and the recommendations provided by Martin-Higueras et al [2021].
Evaluations Following Initial Diagnosis
The following evaluations are recommended to establish the extent of disease and therapeutic needs in an individual diagnosed with PH3:
Kidney imaging for assessment of number and location of stones and presence of nephrocalcinosis
Baseline 24-hour urine collection with measurement of oxalate, calcium, citrate, pH, urine volume, and other components of a supersaturation profile to identify specific risk factors for stones, information that is valuable in guiding treatment
Measurement of plasma oxalate concentration to assess the degree of oxalate overproduction
Assessment of kidney function (estimated glomerular filtration rate [eGFR]) by serum creatinine concentration, blood urea nitrogen, and/or cystatin C concentrations
If chronic kidney disease (CKD stage 3B or higher) is present, evaluation for systemic oxalosis deposits with the following:
Echocardiography for evidence of cardiomyopathy
Complete blood count for evidence of erythropoietin-resistant anemia
Bone imaging for evidence of sclerosis and pathologic fractures due to oxalate osteodystrophy
Retinal examination for retinal oxalate deposition
Consultation with a medical geneticist, certified genetic counselor, or certified advanced genetic nurse to inform affected individuals and their families about the nature,
mode of inheritance, and implications of PH3 in order to facilitate medical and personal decision making
Treatment of Manifestations
There is no cure for PH3.
Medical therapy and pharmacotherapy to reduce urine supersaturation of calcium oxalate are employed to reduce formation of stones and calcium oxalate crystals that can be injurious to the kidney. This treatment should be continued lifelong and includes the following elements:
Maintenance of high oral fluid intake (>2.5 L per m2 body surface area)
Oral administration of an inhibitor of calcium oxalate crystallization. Most individuals are treated with potassium and/or sodium citrate at a daily total of 1-3 mEq/kg per day divided into two or three doses a day.
Individuals with PH3 with active calcium oxalate stone formation who have hypercalciuria or urine calcium that is in the upper range of normal may benefit from the addition of a thiazide medication.
Stone management. Stones impose a large clinical burden, impair quality of life, and recur lifelong in individuals with PH3. Furthermore, symptomatic stones often first occur in infancy or early childhood and present greater challenges for surgical stone removal in very young children. Care is often best provided by collaboration between a urologist and nephrologist, who are experienced in the care of individuals with PH, with the following goals [Carrasco et al 2015]:
Monitor stone-forming activity through regularly scheduled kidney imaging to promptly identify stones that are likely to either (1) cause kidney obstruction and kidney damage or (2) benefit from preemptive intervention to avoid acute, symptomatic events.
Prevent stone complications (any of which can damage kidney function) by:
Promptly alleviating obstruction of the urinary tract by a stone through stent placement and/or stone removal. Stone removal procedures should minimize kidney injury as much as possible. For example, ureteroscopic or percutaneous nephrolithotomy may be preferable to repeated extracorporeal shockwave lithotripsy.
Maintaining continuous fluid intake and urine flow before, during, and for several days after stone removal procedures.
Treating urinary tract infections promptly and thoroughly, as bacteria may cause pyelonephritis or infect stones and complicate management.
Avoid acute kidney injury by preventing intravascular volume contraction at all times. This may necessitate intravenous fluids during severe gastroenteritis or other circumstances in which adequate oral fluid intake cannot be maintained.
Avoid marked dietary oxalate excess. The diet should contain the recommended daily allowance for calcium and should avoid excessive intake of foods high in oxalate. Because dietary calcium binds oxalate in the gut, it prevents overabsorption of free (unbound) oxalate. Information on high-oxalate foods can be found at Oxalosis and Hyperoxaluria Foundation, Mayo Clinic, and Harvard Health Publishing.
Avoid supersaturation of calcium oxalate in the blood. Although there is little experience with CKD in PH3, as in other forms of PH a decline in GFR to less than approximately 40 mL/min per 1.73 m2 would be expected to result in increased plasma oxalate and the potential for systemic oxalosis. If plasma oxalate exceeds 35-50 μmol/L, dialysis or transplantation is needed to reduce the risk of multiorgan complications of calcium oxalate deposition.
Surveillance
Attention to lifelong ongoing care, including adherence to high fluid intake and medication schedule, is essential to favorable outcomes. Individuals with PH3 should be counseled to promptly report stone-related symptoms to their health care provider.
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations in those who are stable and doing well (except very young individuals; see Note), the following are recommended annually:
Clinical assessment of stone-related symptoms including pain, frequency of passage of stones or gravel in the urine, and urinary tract infections
Renal ultrasound examination or other imaging to monitor for stone formation
Note: Given the need for lifelong repeated kidney imaging, care should be taken to minimize radiation exposure.
Assessment of kidney function (serum creatinine and eGFR) and electrolytes
Measurement of plasma oxalate concentration, particularly in individuals with any impairment of GFR
24-hour urine oxalate and supersaturation study. During follow up, changes in the urine supersaturation can be used to monitor the effectiveness of therapy by confirming that the crystallization potential has decreased.
Note: In young children or other individuals unable to complete an accurate 24-hour urine collection, random urine specimens may be used for comparative measurements.
Note: Individuals who need more frequent assessments are:
Children under age four years;
Individuals with complex stone problems;
Individuals with reduced kidney function.
Agents/Circumstances to Avoid
Individuals with PH3 should avoid the following:
Intravascular volume contraction
Note: Liberal use of intravenous fluids is indicated whenever oral fluid intake is inadequate or there is loss of body fluids due to diarrhea or other causes.
Delays in treatment of acute stone episodes
Nephrotoxic agents
High-dose ascorbic acid (more than 250 mg daily)
Marked dietary oxalate excess by moderation of intake of high-oxalate foods
Pregnancy Management
In the few reports available to date, pregnancy outcomes in women with PH3 appear to be similar to those in women with PH1 or PH2 [Miao et al 2022]. Women with all types of PH who have good kidney function prior to and during pregnancy have done well and have had healthy infants [Norby & Milliner 2004, Miao et al 2022].
Nonetheless, mothers with PH3 should be managed as a higher-risk pregnancy with closer monitoring, given the increased risk of acute kidney injury due to hypovolemia or an obstructing or infected stone [Carrasco et al 2015]. Adequate fluid intake should be maintained throughout the pregnancy, especially in circumstances that compromise fluid intake, such as hyperemesis gravidarum, which require prompt initiation of intravenous fluid to maintain adequate hydration.
Stones that become symptomatic during pregnancy may require routine (but specialized) techniques for management.
Urinary tract infections in individuals who have stones should be treated promptly and thoroughly due to the potential complications of pyelonephritis or infected stones.
Therapies Under Investigation
Evidence suggests that inhibition of the hepatic isoform of lactate dehydrogenase, LDHa, could reduce oxalate generation in PH1, PH2, and PH3.
Preclinical studies have also demonstrated the potential for hepatically directed
gene editing of the LDHa enzyme [
Dejban & Lieske 2022].
Stiripentol, a pharmacologic inhibitor of LDH used to treat Dravet syndrome, a rare form of myoclonic epilepsy, is also being investigated as a therapy for PH [Dejban & Lieske 2022]. Other newer pharmacologic inhibitors of LDHa are also under preclinical investigation.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Primary hyperoxaluria type 3 (PH3) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
If a
pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the
proband occurred as a
de novo event in the proband or as a
postzygotic de novo event in a mosaic parent [
Jónsson et al 2017]. If the proband appears to have
homozygous pathogenic variants (i.e., the same two pathogenic variants), additional possibilities to consider include:
Sibs of a proband
If both parents are known to be
heterozygous for a
HOGA1 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic
carrier, and a 25% chance of being unaffected and not a carrier.
Offspring of a proband. Unless an affected individual's reproductive partner also has PH3 or is a carrier, offspring will be obligate heterozygotes (carriers) for a pathogenic variant in HOGA1. Heterozygotes (carriers) are usually asymptomatic [Bar et al 2021].
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a HOGA1 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the HOGA1 pathogenic variants in the family.
Prenatal Testing and Preimplantation Genetic Testing
Once the HOGA1 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Chapter Notes
Author Notes
The Rare Kidney Stone Consortium (RKSC), funded by the Oxalosis and Hyperoxaluria Foundation and the Mayo Foundation, is an affiliate member of the Rare Disease Clinical Research Network of the National Institutes of Health. The Consortium maintains registries for patients with primary hyperoxaluria, enteric hyperoxaluria, cystinuria, Dent disease, and APRT deficiency. A biobank, protocols for genetic testing, and a number of other protocols are open to enrollment for patients with these diseases.
Dr John Lieske and Dr David J Sas are actively involved in clinical research regarding individuals with primary hyperoxaluria 3. They would be happy to communicate with persons who have any questions regarding diagnosis of PH3 or other considerations.
Dr Lieske and Dr Sas are also interested in hearing from clinicians treating families affected by monogenic stone diseases in whom no causative variant has been identified through molecular genetic testing of the genes known to be involved in this group of disorders.
Contact Dr Peter Harris to inquire about review of HOGA1 variants of uncertain significance.
Acknowledgments
NIH grants U54 DK083908, R21 TR03174, and R01 DK133171 and the Oxalosis and Hyperoxaluria Foundation (OHF) have provided funding for our work.
We also thank the OHF for partnership and funding support for the RKSC PH Registry.
We express our appreciation to the dedicated staff of the Mayo Clinic Hyperoxaluria Center and to all the physicians and patients who have contributed to the RKSC PH Registry.
Revision History
9 February 2023 (bp) Comprehensive update posted live
24 September 2015 (me) Review posted live
13 February 2015 (dsm) Original submission