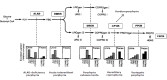
Clinical Description
Variegate porphyria (VP) is classified as both a cutaneous and an acute porphyria. It can present with chronic blistering cutaneous manifestations and/or acute attacks of neurovisceral manifestations that may become chronic.
Cutaneous manifestations. Chronic blistering photosensitivity, typically on the backs of the hands, is the most common manifestation of VP. The lesions result from sun exposure that activates porphyrins and makes the skin fragile and prone to blister formation. Lesions are located on sun-exposed areas, especially the dorsal aspects of the hands and less frequently the face, neck, ears, and lower extremities. Because sun-induced damage is not acute, the role of sunlight is often not recognized. Cutaneous manifestations may improve in winter and be less prevalent in northern regions and in dark-skinned individuals.
These and other manifestations of VP appear typically in adulthood and rarely before puberty.
The subepidermal vesicles, bullae, and erosions crust over and heal slowly. When blisters rupture they may become infected and painful.
Other chronic skin findings include milia, scarring, thickening, and areas of decreased and increased skin pigmentation. Facial hyperpigmentation and hypertrichosis may occur.
The skin manifestations are identical to those seen in porphyria cutanea tarda (PCT) and hereditary coproporphyria (HCP), and less severe than those seen in congenital erythropoietic porphyria (CEP) and hepatoerythropoietic porphyria (HEP). They contrast with the acute non-blistering photocutaneous manifestations of erythropoietic protoporphyria (EPP) (see Table 3).
Of note, the great majority of individuals who are heterozygous for a PPOX pathogenic variant are asymptomatic and are unlikely to be recognized unless they are screened for VP based on a family history of VP (see Genetic Counseling). In South Africa the frequency of acute attacks has decreased in recent decades. This may be due to less common use of harmful drugs such as barbiturates and sulfonamide antibiotics in clinical practice and perhaps better case recognition and better dissemination of information on how to avoid future attacks. VP now more commonly presents in South Africa with cutaneous rather than acute manifestations [Meissner et al 2003, Anderson et al 2005, Hift & Meissner 2005].
Neurovisceral symptoms can occur at any age after puberty as acute attacks, but may become chronic. Symptoms are more common in women than men, and occur less often in the elderly. The frequency and severity of attacks vary considerably and are determined, in part, by exacerbating factors such as certain drugs, hormones, and nutritional deficits [Anderson et al 2005]. The proportion of persons heterozygous for a PPOX pathogenic variant who experience acute attacks decreased from about 30%-40% in the 1980s to 5%-10% in 2005 [Hift & Meissner 2005].
The neurovisceral symptoms are identical to those in the other acute porphyrias (see Differential Diagnosis).
Acute manifestations vary. The most common symptoms are abdominal pain; nausea and vomiting; constipation; pain in the back, chest, and extremities; anxiety; seizures; and a predominantly motor peripheral neuropathy resulting in muscle weakness that may progress to quadriparesis and respiratory paralysis [Kauppinen & Mustajoki 1992, Meissner et al 2003, Anderson et al 2005, Hift & Meissner 2005]. Psychiatric disturbances and autonomic neuropathy can also be observed. Not all symptoms are present in a single episode and symptoms can vary from episode to episode; however, recurrent attacks are often similar. Acute attacks may be severe and are potentially fatal, but on average are less frequent and less severe than those observed in acute intermittent porphyria (AIP) [Hift & Meissner 2005].
Motor neuropathy usually manifests initially as proximal upper-extremity muscle weakness and can be difficult to detect. Hyperreflexia may be seen initially, followed by hyporeflexia as the motor neuropathy progresses. The motor neuropathy may be accompanied by sensory loss. Note: Motor neuropathy due to acute porphyrias is accompanied by little or no elevation of cerebrospinal fluid protein, which helps to differentiate it from the Landry Guillain-Barré syndrome [Anderson et al 2005].
Because abdominal pain is neuropathic rather than inflammatory, abdominal findings are minimal compared to the severity of the pain. Ileus and bladder distension may be present.
An acute attack can be fatal in the presence of severe manifestations including neuropathy, seizures, and respiratory compromise. If managed properly, the outcome of an acute attack is generally good. Even severe motor neuropathy is reversible with recovery over a variable period of months and sometimes over several years.
Factors that predispose to acute attacks that are often identified include exposure to a harmful drug, alcohol, reduced dietary intake, or stress from an infection or other illness. Most harmful drugs are known to be inducers of hepatic δ-aminolevulinic acid synthase (ALAS) and hepatic cytochrome P450 enzymes (see Agents/Circumstances to Avoid). Pregnancy is usually well tolerated but can precipitate acute attacks in some women.
Physical findings such as tachycardia, hypertension, restlessness, and agitation result from autonomic neuropathy and increased circulating catecholamines.
Chronic pain may be a manifestation of VP and other acute porphyrias. Depression may be more difficult to link to the disease. Chronic pain and depression may become important management issues.
Chronic liver abnormalities, particularly mild elevation of serum transaminases, are common. Risks for development of hepatocellular carcinoma and chronic renal disease are increased in VP (as well as in AIP and HCP). Hepatocellular carcinoma may develop, especially after age 50 years in persons with persistent elevations in porphobilinogen and porphyrins.
Note: The speculation that King George III (and perhaps others in the British royal family) had VP has been discounted [Peters 2011].