Retinal Vasculopathy with Cerebral Leukoencephalopathy and Systemic Manifestations
Synonyms: Cerebroretinal Vasculopathy (CRV); Hereditary Endotheliopathy, Retinopathy, Nephropathy, and Stroke (HERNS); Hereditary Systemic Angiopathy (HSA); Hereditary Vascular Retinopathy (HVR); Retinal Vasculopathy with Cerebral Leukodystrophy (RVCL); RVCL-S
Irene de Boer, MD, Nadine Pelzer, MD, PhD, and Gisela Terwindt, MD, PhD.
Author Information and AffiliationsInitial Posting: September 19, 2019.
Estimated reading time: 22 minutes
Summary
Clinical characteristics.
Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations (RVCL-S) is a small-vessel disease that affects highly vascularized tissues including the retina, brain, liver, and kidneys. Age of onset is often between 35 and 50 years. The most common presenting finding is decreased visual acuity and/or visual field defects. Neurologic manifestations may include hemiparesis, facial weakness, aphasia, and hemianopsia. Migraines and seizures are less frequently described. Renal manifestations may include mild-to-moderate increase in serum creatinine and mild proteinuria; progression to end-stage renal disease (ESRD) is uncommon. Hepatic manifestations frequently include mildly elevated levels of alkaline phosphatase and gamma-glutamyltransferase (GGT). Less common findings include psychiatric disorders, hypertension, mild-to-moderate anemia, and Raynaud phenomenon.
Management.
Treatment of manifestations: Retinal vasculopathy may be treated with laser therapy, which may also prevent and slow the progression of visual impairment; macular edema may respond to bevacizumab; ESRD may require renal replacement therapy (including renal transplantation); corticosteroid therapy may be considered for those with cerebral vasogenic edema; standard treatment for glaucoma, hypertension, migraine headaches, seizure disorders, hypothyroidism, anemia, Raynaud phenomenon, and psychiatric disorders.
Surveillance: Ophthalmologic evaluation, blood pressure assessment, renal function tests (serum creatinine, BUN, and urinalysis to include creatinine and protein content), liver function tests (AST, ALT, alkaline phosphatase, GGT, serum albumin), TSH and free T4, and complete blood count annually starting in the fourth decade or as appropriate based on symptoms; annual assessment of cognition and psychiatric manifestations.
Agents/circumstances to avoid: Intravenous tissue-type plasminogen activator therapy for acute ischemic stroke is not warranted, as there is no proof that neurologic manifestations are caused by occluded large blood vessels and the risk of complications is assumed to be higher in affected individuals.
Evaluation of relatives at risk: It is appropriate to evaluate the genetic status of apparently asymptomatic older and younger at-risk relatives of an affected individual by molecular genetic testing of the TREX1 pathogenic variant in the family in order to identify as early as possible those who would benefit from prompt initiation of treatment and preventive measures.
Genetic counseling.
RVCL-S is inherited in an autosomal dominant manner. Most individuals diagnosed with RVCL-S have an affected parent. However, disease onset and severity vary considerably even within the same family. The offspring of an individual with RVCL-S are at a 50% risk of inheriting the TREX1 pathogenic variant. If the pathogenic variant in the family is known, prenatal testing for pregnancies at increased risk for RVCL-S and preimplantation genetic testing are possible; however, such testing for adult-onset disorders is uncommon.
Diagnosis
There are no consensus clinical diagnostic criteria for retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations (RVCL-S).
Suggestive Findings
RVCL-S should be suspected in individuals with the following findings [Stam et al 2016, Pelzer et al 2019].
Major features
Vascular retinopathy typically manifesting as decreased visual acuity and/or visual field defects
Focal and/or global brain dysfunction and brain MRI abnormalities
Focal neurologic signs can include but are not limited to hemiparesis, facial weakness, aphasia, and hemianopsia.
Global brain dysfunction may manifest as progressive cognitive impairment.
Brain MRI abnormalities are restricted to the white matter (see Clinical Characteristics,
Neurologic Features).
Family history of middle-age onset of disease manifestations consistent with an
autosomal dominant inheritance pattern
Note: Absence of a known family history of similarly affected individuals does not preclude the diagnosis.
Supportive features
Calcifications on brain CT scan, typically not present in healthy controls
Nonspecific MRI white matter lesions that occur more frequently than expected given the age of the individual
Microvascular liver disease, manifested by modest elevations of alkaline phosphatase and gamma-glutamyltransferase
Microvascular kidney disease, typically manifested by a mild-to-moderate increase in serum creatinine or by proteinuria
Likely associated features
Anemia consistent with blood loss and/or chronic disease (typically normocytic and normochromic)
Microscopic gastrointestinal bleeding
Hypertension
Migraine with or without aura
Raynaud phenomenon (typically mild)
Subclinical hypothyroidism
Establishing the Diagnosis
The diagnosis of RVCL-S is established in a proband with suggestive findings and a heterozygous pathogenic variant in TREX1 identified by molecular genetic testing (see Table 1).
Molecular genetic testing approaches can include single-gene testing and use of a multigene panel:
A
multigene panel that includes
TREX1 and other genes of interest (see
Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of
uncertain significance and pathogenic variants in genes that do not explain the underlying
phenotype. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused
exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Note: At the Leiden University Medical Center, a panel of genes is used to screen for pathogenic variants that cause cerebral angiopathies and adult-onset leukoencephalopathies. New pathogenic variants that cause these disorders continue to be discovered; click
here for more information.
Table 1.
Molecular Genetic Testing Used in Retinal Vasculopathy with Cerebral Leukoencephalopathy and Systemic Manifestations
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic
Variant 2 Detectable by Method |
|---|
|
TREX1
| Sequence analysis 3 | Estimated >99% 4, 5 |
| Gene-targeted deletion/duplication analysis 6 | Unknown 7 |
- 1.
- 2.
- 3.
- 4.
The identification of a heterozygous pathogenic variant in TREX1 is required for the diagnosis RVCL-S. The proportion of individuals with suggestive features of RVCL-S who do not have an identifiable heterozygous pathogenic variant in TREX1 has not been systematically studied.
- 5.
To date, all pathogenic variants in TREX1 that cause RVCL-S have been frameshift variants in the region encoding the C terminus of the protein.
- 6.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 7.
Clinical Characteristics
Clinical Description
Clinical information about RVCL-S is summarized from the following reports of individuals with molecularly confirmed RVCL-S: Storimans et al [1991], Terwindt et al [1998], Cohn et al [2005], Richards et al [2007], Winkler et al [2008], Mateen et al [2010], Gruver et al [2011], Schuh et al [2014], Dhamija et al [2015], DiFrancesco et al [2015], Stam et al [2016], Vodopivec et al [2016], Carra-Dalliere et al [2017], Hardy et al [2017], and Pelzer et al [2019]. The numerators and denominators in this section are derived from the study by Stam et al [2016], which included 11 affected families and is the largest study to date on RVCL-S.
RVCL-S is a small-vessel disease that systemically affects various highly vascularized organs. Affected individuals develop retinal vasculopathy and neurologic symptoms, in addition to other systemic manifestations including impaired liver and renal function.
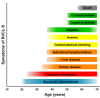
Clinical presentation is variable; onset is often between ages 35 and 50 years, with a mean age at clinical diagnosis of 42.9 years (SD ±8.3, range 25-61 years). Life expectancy is decreased; the average age of death is 53.1 years (SD ±9.6, range 32-72). Cause of death is frequently pneumonia or sepsis in the setting of general debilitation. For clinical course see .
Clinical course of RVCL-S Adapted from Pelzer et al [2019]
Ophthalmologic Features
Symptoms
Symptoms caused by retinal vasculopathy are the most common presenting finding in individuals with RVCL-S but can also develop later in the disease course.
Affected individuals often notice decreased visual acuity and/or visual field defects. A gradual worsening often occurs and affected individuals can become legally blind.
Signs
The retinopathy is characterized in the early stages by telangiectasias, microaneurysms, and cotton wool spots.
In later stages perifoveal capillary obliterations and neovascularizations appear.
Macular edema and neovascular glaucoma may develop as a complication of vascular retinopathy.
Neurologic Features
Symptoms
Focal neurologic symptoms were reported in 40/72 individuals. These symptoms occurred more frequently in those with retinal vasculopathy and/or brain lesions (40/59). Manifestations include but are not limited to hemiparesis, facial weakness, aphasia, and hemianopsia.
Cognitive impairment has been described in 32/57 affected individuals with retinal vasculopathy and/or brain lesions. This ratio is higher (21/28) in those with more disease symptoms, implying a progressive decline in cognition. Apathy, irritability, and difficulties with memory and judgment have also been reported.
Migraine occurred in 24/41 individuals in whom other manifestations of the disease (retinal vasculopathy and/or brain lesions) were present. Of 16 deceased individuals, 12 had a history of migraine at some point in their life; further clinical details are unavailable.
Seizures occurred in 9/66 affected individuals. Generalized as well as partial sensomotoric seizures have been described.
Signs on neuroimaging. Three types of lesions have been observed frequently on brain MRI scans:
Focal, non-enhancing T2-hyperintense lesions scattered throughout the periventricular and deep white matter (at an age when nonspecific age-related white matter hyperintensities are infrequent)
Punctate T2-hyperintense white matter lesions with nodular enhancement
Hyperintense mass lesions on T2 and hypointense lesions on T1-weighted images, enhanced with gadolinium contrast, and often surrounded by extensive edema. Hemorrhages are rarely reported. Occasionally, restricted diffusion, most often centrally, is observed and is referred to as a "pseudotumor." These lesions:
Are most frequently localized in the frontoparietal lobe but are occasionally found in other regions;
Often lead to displacement of adjacent structures and sulci effacement;
Can increase in size, remain stable, or diminish;
Are associated with calcifications on CT scan.
Renal Disease
Renal disease has been demonstrated in 22/44 affected individuals. It is typically characterized by a mild-to-moderate increase in serum creatinine and mild proteinuria but may be severe (stage IV kidney disease) and fatal in some families. While the progression of the renal disease of RVCL-S requires further study, in most cases the renal manifestations are progressive; however, there is tremendous variability in the rate of renal decline.
Liver Disease
Liver disease was present in 28/40 affected individuals and usually manifests as mildly elevated levels of alkaline phosphatase and gamma-glutamyltransferase (GGT).
Hypothyroidism
Subclinical hypothyroidism was found to be part of the disease course in a recent cross-sectional study. Of the 19 affected individuals older than age 40 years, seven had subclinical hypothyroidism.
Psychiatric Symptoms
Psychiatric manifestations were present in 26/62 affected individuals and may include depression, psychosis, anxiety, and other psychiatric problems.
Other
Additional findings may include the following:
Hypertension (present in 30/52 individuals)
Mild-to-moderate anemia that is typically normocytic and normochromic
Microscopic gastrointestinal bleeding resulting in anemia (present in 25/34 affected individuals)
Avascular necrosis of the femoral head (2 individuals)
(Hypertensive) cardiomyopathy (3 individuals)
Macular skin rash and punctate skin lesions (3 individuals)
Pathology
Histologic abnormalities have been demonstrated in all organs involved in RVCL-S, including the following.
Retina
Brain
Multiple – often confluent – foci of ischemic necrosis of white matter
Vasculopathy: vessel wall thickening and luminal stenosis; telangiectasias
A modest chronic inflammatory cell infiltrate in some individuals
Focal calcifications and reactive astrocytosis
Myelin loss
Kidney
Liver
Nodular regenerative hyperplasia
Micro- and macrovesicular steatosis
Periportal inflammation
Bridging and portal fibrosis
Penetrance
Penetrance of RVCL-S is age dependent; however, it is thought that all individuals with a heterozygous pathogenic TREX1 variant will develop features of this condition if they live long enough.
Nomenclature
Previous descriptions of families with cerebroretinal vasculopathy (CRV); hereditary vascular retinopathy (HRV); hereditary endotheliopathy, retinopathy, nephropathy, and stroke (HERNS); hereditary systemic angiopathy (HSA); and retinal vasculopathy with cerebral leukodystrophy (RVCL) represent early reports of RVCL-S.
Prevalence
Currently, fewer than 25 families with RVCL-S are known. However, RVCL-S is most likely underdiagnosed because physicians are generally unfamiliar with the disorder. For example, in the Netherlands alone three unrelated families have been identified because of increased awareness of this condition. The widespread availability of exome and genome sequencing techniques likely also contribute to increased recognition of RVCL-S.
Differential Diagnosis
Due to the systemic nature of retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations (RVCL-S), the differential diagnosis is quite broad. Following are differential diagnoses specifically for the characteristic brain findings and vascular retinopathy seen in RVCL-S. For other inherited disorders with phenotypic similarities see Table 3.
Focal and/or Global Brain Dysfunction / MRI Brain Abnormalities
Focal and/or global brain dysfunction and MRI brain abnormalities (intracerebral mass lesions and white matter abnormalities) seen in RVCL-S can be similar to intracranial neoplasm, multiple sclerosis, multi-infarct dementia, and central nervous system vasculitis. In these disorders other organs are usually not affected. Moreover, these conditions do not typically follow an autosomal dominant inheritance pattern.
In sarcoidosis and systemic lupus erythematosus (SLE) (see
Table 3) there can be focal and/or global brain dysfunction as well as involvement of multiple organs, as is seen in RVCL-S.
With sarcoidosis, however, there is frequent involvement of the lungs and skin. No lesions in the lungs have been described in individuals with RVCL-S and skin lesions are not frequently reported [
Hardy et al 2017].
Vascular Retinopathy
Vascular retinopathy can occur as a long-term complication of diabetes mellitus and hypertension:
As the vascular retinopathy in RVCL-S is usually observed at a relatively young age (retinopathy has been found in the third decade of life) it is unlikely to be a complication of hypertension or diabetes mellitus, even in those with RVCL-S who have co-occurrence of hypertension or diabetes.
Vascular retinopathy can also be caused by systemic lupus erythematosus, sarcoidosis, and ophthalmologic infections (which are outside of the scope of this GeneReview).
In Susac syndrome, vascular retinopathy and encephalopathy are part of the symptomatology, as is hearing loss, but hearing loss is very rare in RVCL-S. The pattern of white matter lesions seen on MRI may also help to distinguish the two conditions.
Table 3.
Inherited Disorders to Consider in the Differential Diagnosis of RVCL-S
View in own window
Differential
Diagnosis
Disorder | Gene(s) | MOI | Clinical Features of the Differential Diagnosis Disorder |
|---|
| Overlapping w/RVCL-S | Distinguishing from RVCL-S |
|---|
|
CADASIL
|
NOTCH3
| AD | Focal neurologic symptoms Progressive cognitive decline Migraine Mood disturbances Apathy White matter lesions on brain imaging Positive family history
|
|
| CARASAL |
CTSA
| AD |
| Hemorrhagic strokes occur frequently in CARASAL. |
|
CARASIL
|
HTRA1
| AR | Stepwise deterioration in brain functions Progressive cognitive decline Extended white matter lesions on brain imaging Positive family history
|
|
|
Fabry disease
|
GLA
| XL |
| Angiokeratomas, hypohidrosis, & corneal opacity Onset usually in childhood or adolescence XL inheritance; heterozygous females can be asymptomatic w/normal life span.
|
|
Neurofibromatosis 1
|
NF1
| AD |
| Café au lait macules, neurofibroma, plexiform neurofibroma, freckling in axillary or inguinal regions, optic glioma, Lisch noduli, & distinctive osseous lesions Pattern of lesions on brain imaging differs from that seen in RVCL-S.
|
Systemic lupus erythematosus
(OMIM 152700) |
CTLA4
DNASE1
FCGR2A
FCGR2B
PTPN22
TREX1
| AD 1 | Nephropathy Anemia Retinopathy Mood disorders Cognitive complaints Seizures Liver disease
| Features of vasculitis can occur, specifically cutaneous abnormalities, oral or nasal ulcers, alopecia, & arthritis. Often associated w/↑ ANAs, anti-dsDNA; anti-Smith antibodies &/or antiphospholipid antibodies (anticardiolipin immunoglobin or lupus anticoagulant)
|
|
Tuberous sclerosis complex
|
TSC1
TSC2
| AD | Multisystem disorder involving brain, kidney, & eye Focal neurologic symptoms Seizures Mass lesions on brain imaging Positive family history
| Formation of hamartomas & skin& lung lesions Many patients are diagnosed as children, but it is not uncommon for adults to be diagnosed.
|
AD = autosomal dominant; ANAs = antinuclear antibodies; AR = autosomal recessive; CADASIL = cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CARASAL = cathepsin A–related arteriopathy with strokes and leukoencephalopathy; CARASIL = cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy; dsDNA = double-stranded DNA; MOI = mode of inheritance; XL = X-linked
- 1.
Most frequently, no genetic cause is identified.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with RVCL-S
View in own window
| System/Concern | Evaluation | Comment |
|---|
|
Eyes
| Ophthalmologic evaluation | Assess for signs of retinopathy, macular edema, & glaucoma. |
|
Cardiovascular
| Blood pressure | Assess for hypertension. |
|
Renal
| Renal function tests 1 | Consider referral to nephrologist for those w/significant renal insufficiency &/or hypertension. |
|
Hepatology
| Liver enzymes 2 & serum albumin Consideration of serum lactate dehydrogenase levels, coagulation studies, & bilirubin concentration
| Consider referral to gastroenterologist for those w/significantly altered liver enzymes or ↓ liver function. |
|
Neurologic
| Assess cognitive function. | Consider referral to neurologist &/or a neuropsychological evaluation. |
| Assess for signs & symptoms of migraines. | Consider referral to neurologist. |
| EEG if seizures are suspected |
| Assess for focal neurologic complaints. | Refer to neurologist, if present. |
|
Endocrinologic
| Thyroid function 3 | Refer to endocrinologist, if clinically significant |
|
Hematologic
| Complete blood count | To assess for anemia; consider referral to gastroenterologist for possible occult GI bleeding. |
|
Rheumatologic
| Assess for signs & symptoms of Raynaud phenomenon. | |
|
Psychiatric
| Assess for psychiatric symptoms. 4 | Consider psychiatric consultation. |
|
Other
| Consultation w/clinical geneticist &/or genetic counselor | |
- 1.
Serum creatinine, BUN, and urinalysis (to include creatinine and protein content)
- 2.
Aspartate transaminase (AST), alanine transaminase (ALT), alkaline phosphatase, gamma-glutamyltransferase (GGT)
- 3.
- 4.
Findings may include depression, psychosis, anxiety, or other psychiatric diagnoses.
It is crucial that unnecessary diagnostic tests not be undertaken. Biopsies of the brain, kidney, or liver are not required in individuals with RVCL-S and do not appear to provide additional prognostic or treatment information.
Treatment of Manifestations
Table 5 summarizes treatment for the manifestations of an individual diagnosed with RVCL-S.
Table 5.
Treatment of Manifestations in Individuals with Retinal Vasculopathy with RVCL-S
View in own window
| Manifestation/Concern | Treatment | Considerations/Other |
|---|
|
Visual impairment
| Retinal laser therapy | Can prevent & slow down progression of retinal involvement that causes visual impairment |
|
Macular edema
| Bevacizumab | |
Elevated eye pressure /
Glaucoma
| Standard treatment | |
|
Hypertension
| Standard treatment | May prevent further damage to involved organs |
|
Renal insufficiency
| Standard treatment of hypertension | Refer to nephrologist. |
| Renal replacement therapy (incl transplantation) may be considered in severe cases for those w/end-stage renal disease. |
|
Liver function abnormalities
| There is no treatment for liver function abnormalities. | Monitor to prevent complications associated w/liver fibrosis & to exclude other causes. 1 |
Cerebral vascular
edema
| Consider corticosteroid therapy. 2 | May reduce cerebral vasogenic edema but does not appear to treat the underlying lesion |
|
Migraine
| Standard treatment | |
|
Seizure disorder
| Standard treatment w/antiepileptic drugs | Refer to neurologist. |
|
Hypothyroidism
| Thyroid replacement therapy | |
|
Anemia
| Standard treatment | Intravenous iron therapy (& in rare cases blood transfusion) may be necessary. |
|
Raynaud phenomenon
| Standard treatment | |
|
Psychiatric concerns
| Standard treatment | Refer to psychiatrist or neuropsychologist. |
- 1.
When not properly monitored, affected individuals are frequently given incorrect diagnoses and often receive advice to stop drinking alcohol or stop their medication, while this is not required.
- 2.
Intravenous methylprednisolone followed by an oral corticosteroid treatment
Surveillance
There are currently no consensus guidelines for monitoring individuals with RVCL-S. However, close monitoring is recommended. The frequency of monitoring depends on the manifestations and rate of disease progression (see Table 6).
Table 6.
Recommended Surveillance for Individuals with RVCL-S
View in own window
| System/Concern | Evaluation | Frequency (Minimum) |
|---|
|
Eyes
| Ophthalmologic evaluation | Annually starting in 4th decade or as appropriate based on symptoms |
|
Cardiovascular
| Blood pressure evaluation |
|
Renal
| Renal function tests 1 |
|
Endocrinologic
| TSH and free T4 |
|
Hematologic
| Complete blood count |
|
Hepatology
| Liver enzymes 2 & serum albumin | Annually starting in the 4th decade |
|
Neurologic
| Assessment of cognition | Annually or as appropriate based on symptoms starting after diagnosis |
|
Psychiatric
| Assessment for psychiatric symptoms |
- 1.
Serum creatinine, BUN, and urinalysis (to include creatinine and protein content)
- 2.
Aspartate transaminase (AST), alanine transaminase (ALT), alkaline phosphatase, gamma-glutamyltransferase (GGT)
Agents/Circumstances to Avoid
Although there is no evidence, the authors would advise against intravenous tissue-type plasminogen activator (IV tPA) for acute ischemic stroke. There is to date no proof that the neurologic manifestations are caused by occluded large blood vessels (RVCL-S is a small vessel disease), and the risk of complications of such treatment (e.g., a secondary hemorrhage) is assumed to be higher in affected individuals.
Evaluation of Relatives at Risk
It is appropriate to evaluate the genetic status of apparently asymptomatic older and younger at-risk relatives of an affected individual by molecular genetic testing of the TREX1 pathogenic variant in the family in order to identify as early as possible those who would benefit from prompt initiation of treatment that could benefit eyesight and prevent secondary damage due to hypertension. Furthermore, clarification of the genetic status of at-risk relatives could prevent unnecessary diagnostic tests (e.g., liver/kidney/brain biopsies).
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations (RVCL-S) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
The absence of clinical manifestations in parents whose genetic status is unknown cannot be used to predict risk to sibs of a
proband because of the possibility of reduced
penetrance or late onset in a
heterozygous parent or the theoretic possibility of parental
germline mosaicism.
Offspring of a proband. Each child of an individual with RVCL-S has a 50% chance of inheriting the TREX1 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the TREX1 pathogenic variant, his or her family members may be at risk.
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with an autosomal dominant condition has the pathogenic variant identified in the proband or clinical evidence of the disorder, the pathogenic variant is likely de novo. However, non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) and undisclosed adoption could also be explored.
Prenatal Testing and Preimplantation Genetic Testing
Once the TREX1 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
RVCL Association
Netherlands
RVCLresearch.org
European Leukodystrophy Association (ELA)
Phone: 03 83 30 93 34
Retina International
Ireland
Phone: 353 1 961 9259
Email: info@retina-International.org
United Leukodystrophy Foundation
Phone: 800-SAV-LIVE; 815-748-3211
Email: office@ulf.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Retinal Vasculopathy with Cerebral Leukoencephalopathy and Systemic Manifestations: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
OMIM Entries for Retinal Vasculopathy with Cerebral Leukoencephalopathy and Systemic Manifestations (View All in OMIM)
View in own window
|
192315 | VASCULOPATHY, RETINAL, WITH CEREBRAL LEUKOENCEPHALOPATHY AND SYSTEMIC MANIFESTATIONS; RVCLS |
|
606609 | 3-PRIME @REPAIR EXONUCLEASE 1; TREX1 |
Gene structure. The TREX1 transcript NM_033629.5 has two exons, the first being a short noncoding exon. Alternative splicing results in alternate transcripts. For a detailed summary of gene, transcript, and protein information see Table A.
Pathogenic variants. In RVCL-S, the sequence alterations in TREX1 are frameshift variants typically resulting in a truncated C terminus of the TREX1 protein [Richards et al 2007, Schuh et al 2014, Dhamija et al 2015, Stam et al 2016, Vodopivec et al 2016, Carra-Dalliere et al 2017].
Normal gene product. TREX1 is a major 3'-5' exonuclease in mammals and is responsible for the removal of nucleoside monophosphates from the 3' ends of DNA [Mazur & Perrino 1999]. It has a high affinity for single-stranded DNA [Höss et al 1999]. TREX1 (NP_338599.1) contains 314 amino acid residues, which are highly conserved among species [Lee-Kirsch et al 2007]. TREX1 has three exonuclease domains in the N terminus and a C-terminal domain, which is required for its localization to the endoplasmic reticulum [Mazur & Perrino 1999, Richards et al 2007, DiFrancesco et al 2015, Hasan et al 2015]. TREX1 also appears to be involved in the regulation of oligosaccharyltransferase activity [Hasan et al 2015].
Abnormal gene product. Heterozygous frameshift pathogenic variants in the C terminus of TREX1 cause RVCL-S [Richards et al 2007, Stam et al 2016]. These are predicted to result in a truncated TREX1 protein. The exonuclease function of the protein is not affected but its subcellular localization is altered [Richards et al 2007, DiFrancesco et al 2015, Hasan et al 2015]. Recently it was demonstrated that the C terminus of TREX1 has a role in the regulation of the oligosaccharlytransferase complex located on the endoplasmic reticulum [Hasan et al 2015]. In lymphoblasts of individuals with RVCL-S this led to an increased free glycan release that could potentially cause immune activation [Hasan et al 2015]. In light of these findings, it is especially interesting that a distinct antibody profile was demonstrated in a mouse model for RVCL-S [Sakai et al 2017]. A remaining question is how pathogenic variants in the C terminus of TREX1 cause the multisystem vasculopathy seen in individuals with RVCL-S [Stam et al 2009].
References
Literature Cited
Carra-Dalliere C, Ayrignac X, Prieto-Morin C, Girard P, Tournier-Lasserve E, Labauge P. TREX1 mutation in leukodystrophy with calcifications and persistent gadolinium-enhancement.
Eur Neurol. 2017;77:113–4. [
PubMed: 28013302]
Cohn AC, Kotschet K, Veitch A, Delatycki MB, McCombe MF. Novel ophthalmological features in hereditary endotheliopathy with retinopathy, nephropathy and stroke syndrome.
Clin Exp Ophthalmol. 2005;33:181–3. [
PubMed: 15807828]
Dhamija R, Schiff D, Lopes MB, Jen JC, Lin DD, Worrall BB. Evolution of brain lesions in a patient with TREX1 cerebroretinal vasculopathy.
Neurology. 2015;85:1633–4. [
PubMed: 26527794]
DiFrancesco JC, Novara F, Zuffardi O, Forlino A, Gioia R, Cossu F, Bolognesi M, Andreoni S, Saracchi E, Frigeni B, Stellato T, Tolnay M, Winkler DT, Remida P, Isimbaldi G, Ferrarese C. TREX1 C-terminal frameshift mutations in the systemic variant of retinal vasculopathy with cerebral leukodystrophy.
Neurol Sci. 2015;36:323–30. [
PubMed: 25213617]
Gruver AM, Schoenfield L, Coleman JF, Hajj-Ali R, Rodriguez ER, Tan CD. Novel ophthalmic pathology in an autopsy case of autosomal dominant retinal vasculopathy with cerebral leukodystrophy.
J Neuroophthalmol. 2011;31:20–4. [
PubMed: 21131853]
Hardy TA, Young S, Sy JS, Colley AF, Terwindt GM, Ferrari MD, Hayes MW, Hodgkinson S. Tumefactive lesions in retinal vasculopathy with cerebral leucoencephalopathy and systemic manifestations (RVCL-S): a role for neuroinflammation?
J Neurol Neurosurg Psychiatry. 2017. Epub ahead of print. [
PubMed: 28794152]
Hasan M, Fermaintt CS, Gao N, Sakai T, Miyazaki T, Jiang S, Li QZ, Atkinson JP, Morse HC 3rd, Lehrman MA, Yan N. Cytosolic nuclease TREX1 regulates oligosaccharyltransferase activity independent of nuclease activity to suppress immune activation.
Immunity. 2015;43:463–74. [
PMC free article: PMC4575271] [
PubMed: 26320659]
Hughes M, Little J, Herrick AL, Pushpakom S, Byers H, Worthington J, Newman WG. A synonymous variant in TREX1 is associated with systemic sclerosis and severe digital ischaemia.
Scand J Rheumatol. 2017;46:77–8. [
PubMed: 27574969]
Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hübner N. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus.
Nat Genet. 2007;39:1065–7. [
PubMed: 17660818]
Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, Perrino FW. The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease.
J Biol Chem. 2008;283:31649–56. [
PMC free article: PMC2581595] [
PubMed: 18805785]
Mateen FJ, Krecke K, Younge BR, Ford AL, Shaikh A, Kothari PH, Atkinson JP. Evolution of a tumor-like lesion in cerebroretinal vasculopathy and TREX1 mutation.
Neurology. 2010;75:1211–3. [
PMC free article: PMC3013489] [
PubMed: 20876473]
Mazur DJ, Perrino FW. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3'-->5' exonucleases.
J Biol Chem. 1999;274:19655–60. [
PubMed: 10391904]
Pelzer N, Hoogeveen ES, Haan J, Bunnik R, Poot CC, van Zwet EW, Inderson A, Fogteloo AJ, Reinders MEJ, Middelkoop HAM, Kruit MC, van den Maagdenberg AMJM, Ferrari MD, Terwindt GM. Systemic features of retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations: a monogenic small vessel disease.
J Intern Med. 2019;285:317–32. [
PubMed: 30411414]
Pelzer N, de Vries B, Boon EM, Kruit MC, Haan J, Ferrari MD, van den Maagdenberg AM, Terwindt GM. Heterozygous TREX1 mutations in early-onset cerebrovascular disease.
J Neurol. 2013;260:2188–90. [
PubMed: 23881107]
Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation.
Nat Genet. 2016;48:126–33. [
PMC free article: PMC4731925] [
PubMed: 26656846]
Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-Labarca ML, Terwindt GM, Kasai Y, McLellan M, Grand MG, Vanmolkot KR, de Vries B, Wan J, Kane MJ, Mamsa H, Schäfer R, Stam AH, Haan J, de Jong PT, Storimans CW, van Schooneveld MJ, Oosterhuis JA, Gschwendter A, Dichgans M, Kotschet KE, Hodgkinson S, Hardy TA, Delatycki MB, Hajj-Ali RA, Kothari PH, Nelson SF, Frants RR, Baloh RW, Ferrari MD, Atkinson JP. C-terminal truncations in human 3'-5' DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy.
Nature genetics. 2007;39:1068–70. [
PubMed: 17660820]
Sakai T, Miyazaki T, Shin DM, Kim YS, Qi CF, Fariss R, Munasinghe J, Wang H, Kovalchuk AL, Kothari PH, Fermaintt CS, Atkinson JP, Perrino FW, Yan N, Morse HC 3rd. DNase-active TREX1 frame-shift mutants induce serologic autoimmunity in mice.
J Autoimmun. 2017;81:13–23. [
PMC free article: PMC5558601] [
PubMed: 28325644]
Schuh E, Ertl-Wagner B, Lohse P, Wolf W, Mann JF, Lee-Kirsch MA, Hohlfeld R, Kümpfel T. Multiple sclerosis-like lesions and type I interferon signature in a patient with RVCL.
Neurol Neuroimmunol Neuroinflamm. 2014;2:e55. [
PMC free article: PMC4277301] [
PubMed: 25566545]
Stam AH, Haan J, van den Maagdenberg AM, Ferrari MD, Terwindt GM. Migraine and genetic and acquired vasculopathies.
Cephalalgia. 2009;29:1006–17. [
PubMed: 19689610]
Stam AH, Kothari PH, Shaikh A, Gschwendter A, Jen JC, Hodgkinson S, Hardy TA, Hayes M, Kempster PA, Kotschet KE, Bajema IM, van Duinen SG, Maat-Schieman ML, de Jong PT, de Smet MD, de Wolff-Rouendaal D, Dijkman G, Pelzer N, Kolar GR, Schmidt RE, Lacey J, Joseph D, Fintak DR, Grand MG, Brunt EM, Liapis H, Hajj-Ali RA, Kruit MC, van Buchem MA, Dichgans M, Frants RR, van den Maagdenberg AM, Haan J, Baloh RW, Atkinson JP, Terwindt GM, Ferrari MD. Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations.
Brain. 2016;139:2909–22. [
PMC free article: PMC5091044] [
PubMed: 27604306]
Storimans CW, Van Schooneveld MJ, Oosterhuis JA, Bos PJ. A new autosomal dominant vascular retinopathy syndrome.
Eur J Ophthalmol. 1991;1:73–8. [
PubMed: 1821204]
Terwindt GM, Haan J, Ophoff RA, Groenen SM, Storimans CW, Lanser JB, Roos RA, Bleeker-Wagemakers EM, Frants RR, Ferrari MD. Clinical and genetic analysis of a large Dutch family with autosomal dominant vascular retinopathy, migraine and Raynaud's phenomenon.
Brain. 1998;121:303–16. [
PubMed: 9549508]
Winkler DT, Lyrer P, Probst A, Devys D, Haufschild T, Haller S, Willi N, Mihatsch MJ, Steck AJ, Tolnay M. Hereditary systemic angiopathy (HSA) with cerebral calcifications, retinopathy, progressive nephropathy, and hepatopathy.
J Neurol. 2008;255:77–88. [
PubMed: 18204807]
Chapter Notes
Author Notes
Leiden University Medical Center web pages (in Dutch)
RVCL-S patient information
RVCL research
The Leiden University Medical Center has expertise in the diagnosis and treatment of RVCL-S and other cerebral hereditary angiopathies (CHA). As part of a consortium we were involved in finding the locus and gene – TREX1 – responsible for RVCL-S. We aim to study RVCL-S disease pathology through our collaboration with world-renowned scientists and our integrated clinical, radiologic, molecular, and biological approach with this unique patient group and our transgenic mouse model, and aim to include new exciting research opportunities such as brain-on-chip development.