Clinical Description
SCN3A-related neurodevelopmental disorder (SCN3A-ND) encompasses a spectrum of clinical severity associated with epilepsy and/or malformation of cortical development (MCD). Affected individuals may have developmental and epileptic encephalopathy (DEE) (i.e., intractable seizures with developmental delays associated with ongoing epileptiform EEG activity) with or without MCD, or MCD with or without mild focal epilepsy.
To date, 38 individuals from 31 families have been identified with a pathogenic variant in SCN3A [Iossifov et al 2014, Lamar et al 2017, Trujillano et al 2017, Miyatake et al 2018, Smith et al 2018, Tumienė et al 2018, Zaman et al 2018, Li et al 2019, Inuzuka et al 2020, Zaman et al 2020, Ziats et al 2020]. The following summarizes the clinical findings in these individuals.
Table 2.
Select Features of SCN3A-Related Neurodevelopmental Disorder
View in own window
| Feature | Proportion of
Persons w/Feature | Comment |
|---|
|
Developmental delay
| 38/38 (100%) | Severity ranges widely from mild isolated speech delay to profound developmental delays. |
|
Malformation of cortical development
| 31/38 (82%) | |
|
Epilepsy
| 26/38 (68%) | A common but not obligatory feature |
|
Autonomic dysregulation
| 40%-60% | Features typically assoc w/DEE |
|
Infantile hypotonia
| 16/30 (53%) |
|
Oromotor dysfunction
| 13/32 (41%) | Most prominent in those w/o DEE |
|
Progressive microcephaly
| 12/30 (40%) | Features typically assoc w/DEE |
|
Hyperkinetic movement disorder
| 6/33 (18%) |
DEE = developmental and epileptic encephalopathy
SCN3A-ND represents a clinical spectrum in which three main phenotypes are described:
Note: One known affected individual was a fetus whose clinical features could not be further categorized. Two additional individuals with pathogenic SCN3A variants could not be categorized because of incomplete clinical and neuroimaging data [Iossifov et al 2014, Zaman et al 2020].
Because the small number of recognized affected individuals with each phenotype and the fact that the clinical overlap between these phenotypes is broad, the following discussion applies to all three phenotypes.
Developmental delay (DD) / intellectual disability (ID). Some degree of early childhood DD is seen in all affected individuals – although the severity varies widely, ranging from isolated speech delay to severe developmental delay.
In those with intractable seizures with ongoing epileptiform EEG activity:
All affected individuals have severe-to-profound developmental delays and are nonverbal and nonambulatory.
Ongoing ID is present in older individuals, ranging from severe to profound.
In those who have mild focal epilepsy or no epilepsy, long-term cognitive outcomes include:
Normal cognition in 2/11 (18%)
Borderline/mild ID in 7/11 (64%)
Moderate/severe ID in 2/11 (18%)
Neurologic. Most affected individuals have infantile hypotonia, although this may be absent or mild in those who have mild focal epilepsy or no epilepsy.
In individuals with DEE, neurologic examination typically reveals significant truncal or generalized hypotonia with prominent head lag in younger children.
In those with malformation of cortical development:
Hypotonia often progresses to spastic quadriplegia in later childhood in those who also have DEE;
In those without DEE, older individuals may have pyramidal signs, mild spasticity, or mild hemiparesis.
Epilepsy. The type and course of seizures are variable, and depend on whether an affected individual has DEE and on the presence or absence of a malformation of cortical development.
In those with DEE:
Seizure onset is typically in the first six to 12 months of life, most often in the first week of life (median age 1-2 weeks), although seizure onset can range from day of birth to age five years.
The most common presenting seizure types in those without MCD are generalized tonic seizures or epileptic spasms. In those with MCD, the most common presenting seizure types are generalized tonic and focal autonomic seizures.
Tonic seizures may have a prominent autonomic or apneic component, consistent with apparent life-threatening events (ALTEs) in infants.
Affected individuals typically develop additional seizure types, which can include the following:
Seizures remain intractable to multiple anti-seizure medications in approximately 50% of individuals without MCD and in 90% of individuals with MCD.
In those with MCD without DEE:
A majority (11/13; 85%) do not have a history of seizures or epilepsy;
A minority (2/13; 15%) have a single unprovoked focal or generalized tonic-clonic seizure in late childhood or adolescence not requiring anti-seizure medication.
EEG findings. The DEEs include many defined syndromes characterized by a range of seizure types and EEG findings. Interictal EEG patterns include diffuse, multifocal, or focal slowing; generalized or multifocal epileptiform discharges; burst-suppression seen in Ohtahara syndrome; attenuation/low voltage; generalized polyspike-wave or slow spike-wave discharges; hypsarrhythmia as seen in association with infantile spasms syndrome; and others. EEG findings seen in SCN3A-ND may include:
Multifocal epileptiform discharges or hypsarrhythmia in those with DEE but without MCD;
Burst-suppression early in the disease course, then multifocal epileptiform discharges, and hypsarrhythmia in those with MCD;
Focal epileptiform abnormalities on EEG even in the absence of clinical seizures in 70% (10/13) of reported individuals with SCN3A-ND with MCD without DEE.
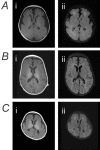
Brain MRI findings can vary from normal to showing thinning or hypoplasia of the corpus callosum to various malformations of cortical development (see Suggestive Findings, Brain MRI findings and ) [Smith et al 2018, Zaman et al 2020]. As some individuals with severe DEE are without MCD, the presence and extent of MCD does not appear to correlate with clinical phenotype. However, diffuse dysgyria is typically associated with severe/profound DEE (14/15; 93%), while focal unilateral or bilateral perisylvian polymicrogyria (PMG) is most often seen in those with a more mild phenotype ranging from normal neurocognitive function to mild/moderate ID, and no history or seizures or infrequent seizures only (11/13; 85%).
Magnetic resonance imaging scans of individuals with SCN3A-related neurodevelopmental disorder A. MRI of the brain of a person with profound global developmental delay and treatment-resistant epilepsy at age two years who had a de novo heterozygous p.Ile875Thr (more...)
Autonomic dysregulation. Ictal and/or non-ictal autonomic disturbances are observed in 45% (9/20) of individuals with DEE [Lamar et al 2017, Zaman et al 2020]. Features include episodes of the following:
Asymmetric or unilateral flushing / color change of the face, which may be confused with Harlequin syndrome
Brady- or tachycardia
Apnea and cyanosis with oxygen desaturation
Excessive sweating
Anisocoria with sluggish pupillary response to light
These episodes may occur several times per day and may or may not be associated with ictal abnormalities on EEG.
Oropharyngeal/Feeding
Oromotor dysfunction is seen in a majority of individuals [
Smith et al 2018,
Zaman et al 2020], and represents a prominent component of the clinical presentation in those with
SCN3A-ND who have bilateral perisylvian PMG. Clinical features may include the following:
Abnormal tongue movements
Speech/language difficulties, including dysarthria
Difficulties whistling, blowing
Abnormal brisk jaw jerk
Some affected individuals with the severe
SCN3A-ND
phenotype exhibit chronic failure to thrive with G-tube dependence for feeding.
Other associated features in some individuals with DEE: