Clinical Description
Spastic paraplegia 15 (SPG15), a form of early-onset complex hereditary spastic paraplegia, is characterized by progressive spasticity that begins in the lower extremities and is associated with several manifestations resulting from central and peripheral nervous system dysfunction (Table 2). Dysfunction of other organ systems has not been established. Onset is typically in mid- to late childhood or adolescence (i.e., between ages 5 and 18 years), though subtle manifestations, such as developmental delay or learning disability, may be present earlier and often precede motor involvement. Individuals with adult onset have been reported. Though natural history data are currently not available, SPG15 is thought to be a progressive disorder. The oldest reported individuals with SPG15 are adults.
The following clinical description of SPG15 is based on a review of more than 70 individuals with biallelic pathogenic variants in ZFYVE26 [Boukhris et al 2008; Hanein et al 2008; Denora et al 2009; Goizet et al 2009; Schüle et al 2009; Schicks et al 2011; Yoon et al 2013; Mallaret et al 2014; Pensato et al 2014; Renvoisé et al 2014; Kara et al 2016; Vinci et al 2018; Özdemir et al 2019; Pascual et al 2019; Araujo et al 2020; Authors, clinical experience].
Of note, some clinical features may present or progress in an age-dependent manner.
Table 2.
Spastic Paraplegia 15: Frequency of Select Features
View in own window
| Feature | % of Persons
w/Feature | Comment |
|---|
| Spasticity | >95% | Lower extremities are involved first & more severely than upper extremities. |
| Pyramidal signs | >95% | Babinski sign, hyperreflexia, ankle clonus |
| Cognitive impairment | >85% | Intellect ranges from normal, learning disability, intellectual disability (often mild to moderate) to progressive cognitive decline. |
| Cerebellar dysfunction | ~55% | Ataxia, dysarthria, nystagmus |
| Dysarthria | ~55% | Bulbar/cerebellar |
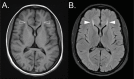
| White matter signal changes | >50% | Most commonly involving periventricular white matter, incl the ears of the lynx sign |
| Peripheral neuropathy & distal amyotrophy | ~40% | Axonal motor & sensory neuropathy on nerve conduction studies; loss of distal muscle bulk, esp in lower extremities |
| Extrapyramidal movement disorder | ~25% | Dystonia (often focal), parkinsonism (variable response to L-dopa) |
|
Pes cavus
| ~20% | |
| Neurogenic bladder dysfunction | ~20% | Urinary urgency, incontinence |
| Pigmentary retinopathy | ~15% | Variable & possibly underdiagnosed (may go unnoticed clinically); part of Kjellin syndrome |
Spasticity. Many individuals have a history of delayed motor milestones. First reported manifestations are often poor balance, clumsiness, and gait impairment, typically in mid- to late childhood. Over time these findings evolve into progressive lower extremity weakness and spasticity with associated pyramidal signs (extensor plantar reflex, hyperreflexia, ankle clonus).
Many individuals, over the course of years, become nonambulatory and ultimately require mobility aids or a wheelchair.
Spasticity progresses to involve the upper extremities in some cases, resulting in a spastic tetraplegia, but continues to be more severe in the legs.
Associated complications may include dysphagia, contractures secondary to progressive spasticity, scoliosis, foot deformities, and dysregulation of bladder and bowel function.
Cognitive impairment. Many individuals experience delayed speech development and/or a learning disability. The degree of cognitive impairment associated is variable and ranges from learning disabilities to mild or moderate intellectual disability. Normal cognition has also been reported. A subset of individuals shows cognitive decline with disease progression.
Cerebellar dysfunction ranges from dysarthria, dysmetria, dysdiadochokinesia, intention tremor, and nystagmus to cerebellar ataxia.
Dysarthria of bulbar and/or cerebellar origin often develops along with the spastic paraplegia.
White matter signal changes (see Suggestive Findings). The most common neuroimaging findings:
Thinning of the corpus callosum
Signal abnormalities of the periventricular white matter
Cerebral and/or cerebellar atrophy
Peripheral neuropathy. With disease progression there is often a loss of muscle bulk, particularly in the distal lower extremities. Nerve conduction studies may show an axonal sensorimotor neuropathy in a subset of affected individuals.
Extrapyramidal movement disorder. A subset of individuals may present with extrapyramidal movement disorders, which may include focal dystonia, often of the limbs, or parkinsonism with bradykinesia, rigidity, tremor, and postural instability.
Pigmentary retinopathy, classically described as part of Kjellin syndrome, may be present in a subset of individuals and is likely underdiagnosed as overt deficits may not be present early in the disease course. Clinical manifestations of the retinopathy, such as defective dark adaptation, color vision loss, changes in visual fields, and central vision loss, are difficult to establish since most affected individuals have intellectual disability and often are nonverbal. Early-onset cataracts have also been reported.
Other
Prevalence
SPG15 is rare. To date about 75 individuals have been reported.
Families with SPG15 have been reported from North America, Europe, the Middle East, Indian subcontinent, East Asia, and South America. Many affected individuals have a history of consanguinity; however, this could be the result of ascertainment bias, as initial reports have mainly focused on families from countries with high rates of consanguinity. More recently, SPG15 has been reported in populations with low rates of consanguinity, often associated with compound heterozygous variants.