Affected Males
To date, information on about 200 individuals with a pathogenic variant in SLC16A2 has been published [Groeneweg et al 2019, Remerand et al 2019]. The following description of the phenotypic features associated with this condition is based on the report by Remerand et al [2019].
Table 2.
Select Features of Allan-Herndon-Dudley Syndrome in Affected Males
View in own window
| Feature 1 | % of Persons w/Feature |
|---|
Prenatal/
neonatal
findings
| Weak fetal movements | 1.4%-16.6% |
| Fetal arrhythmia | 0%-1.4% |
| Neonatal hypotonia | 4.4%-9.4% |
| Premature birth | 0%-0.7% |
| Neonatal hypotrophy | 0%-4.2% |
| Congenital microcephaly | 0%-0.7% |
| Congenital macrocephaly | 0%-0.7% |
| Hydramnios | 0%-1.4% |
| Neonatal jaundice | 0%-20.8% |
|
Growth
| Weight gain deficiency | 33.3% |
| Low weight | 37%-66.6% |
| Short stature | 12.6%-29.1% |
| Microcephaly | 10%-33.3% |
|
DD/ID
| ID | 100% 2 |
| Severe-to-profound ID | 37.5%-83.3% 2 |
| Mild-to-moderate ID | 16.6%-62.5% 2 |
| Oral language | 19.9%-69% |
| Walking | 19.9%-62% |
|
Neuromuscular
| Axial hypotonia | 74%-100% |
| Amyotrophy | 34.5%-88% |
| Spasticity/hyperreflexia | 70.8%-94% |
| Dystonia | 0%-75% |
| Choreoathetosis | 0%-50% |
| Paroxysms or kinesigenic dyskinesias | 0%-9% |
| Ataxia | 0%-60% |
| Seizures | 14.8%-29.1% |
| Nystagmus | 0%-16.6% |
|
Skeletal
| Pectus excavatum | 9.1%-58% |
| Kyphoscoliosis | 21.1%-53.0% |
| Flat feet with valgus | 4.3%-77% |
|
Other
| Narrow/elongated myopathic face | 31%-75% |
| Cryptorchidism | 2.8%-33.3% |
| Peripheral dysthyroidism | 27.9%-66.6% |
|
Brain MRI
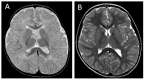
| Severely delayed myelination | 33.1%-79.1% |
| Myelination improvement | 8.4%-62.4% |
| Brain atrophy | 17%-41.6% |
DD = developmental delay; ID = intellectual disability
- 1.
Features and percentages of persons with feature were evaluated from the cohorts of Schwartz et al [2005], Remerand et al [2019], and the entire literature reporting persons with AHDS. Variation in percentages can be attributed to either the non-evaluation or lack of systematic evaluation of features in different reports.
- 2.
Expressed as % of all males with AHDS. All affected persons had ID ranging from mild to profound.
Prenatal/neonatal findings. Infants with AHDS have normal length, weight, and head circumference at birth. Hypotonia, feeding difficulties and early weight gain deficiency can appear in the first weeks or months of life. Prolonged neonatal jaundice has recently been reported.
Growth. Weight gain lags behind linear growth; low weight is a frequent feature Linear growth is frequently normal initially, but between 10 and 30% of males with time have short stature; microcephaly becomes apparent with age.
Developmental delay / intellectual disability. Most affected males have profound-to-severe intellectual disability with no acquisition of walking; most affected males never speak or may develop only garbled sounds secondary to severely dysarthric speech.
Less frequently, affected males have mild-to-moderate intellectual disability, and develop the ability to walk (with or without aid) and use of language allowing academic learning with aid.
Neuromuscular. Truncal hypotonia, a main feature of AHDS, persists into adulthood. Adults are described with "limber neck" or poor head control.
Progressive hypertonicity of the limbs with brisk reflexes, ankle clonus, and extensor plantar responses (Babinski sign) leads to spastic quadriplegia and joint contractures.
Overall muscle mass (particularly proximally) is reduced and associated with generalized muscle weakness.
It is common for affected males to experience purposeless movements described as dystonic and/or athetoid and characteristic paroxysms or kinesigenic dyskinesias [Brockmann et al 2005, Fuchs et al 2009]. These can be triggered by somatosensory stimuli, including changing clothes or diaper, or lifting the affected child. During attacks, the body extends and the mouth opens; stretching or flexing of the limbs lasts as long as one to two minutes.
Some authors also reported abnormal movements as ataxia [Schwartz et al 2005].
Seizures typically begin during infancy or early childhood. Drug resistance is common [Schwartz & Stevenson 2007, Remerand et al 2019].
Rotary nystagmus and disconjugate eye movements have been reported but are not common [Dumitrescu et al 2004, Remerand et al 2019].
Skeletal. Pectus excavatum and kyphoscoliosis are most likely the result of hypotonia and reduced muscle mass.
Behavior. Generally, affected individuals are attentive, friendly, and docile. They are not aggressive or destructive.
Other. Peripheral dysthyroidism can be expressed as cold intolerance, sweating, intestinal transit disorders, tachycardia, high blood pressure, and sleep disorders.
Life span. Early death has occurred in some individuals, usually caused by recurrent infections and/or aspiration pneumonia. In a few instances survival beyond age 70 years has been reported.