Clinical Description
TNXB-related classical-like Ehlers-Danlos syndrome (clEDS) was first reported in 1997 [Burch et al 1997]. It was noted that the clinical features of these individuals strongly resembled classic EDS (cEDS) because of hyperextensible skin and generalized hypermobility, with two key differences: (1) absence of atrophic scarring and (2) autosomal recessive inheritance. Due to the clinical resemblance with cEDS, updated nosology has renamed the condition classical-like EDS (clEDS) [Malfait et al 2017]. Since the first publications, other features have been reported in individuals with clEDS that are more specific to clEDS [Brady et al 2017, Demirdas et al 2017]. These are reflected in the minor criteria for clEDS and include broad feet and hands, brachydactyly, edema in the legs in the absence of cardiac failure, and predisposition to tissue fragility, particularly of the gastrointestinal tract [Green et al 2020].
To date, 56 individuals from 44 families have been identified with TNXB-related clEDS [Burch et al 1997, Schalkwijk et al 2001, Lindor & Bristow 2005, Voermans et al 2007, Voermans et al 2009, Besselink-Lobanova et al 2010, O'Connell et al 2010, Hendriks et al 2012, Pénisson-Besnier et al 2013, Sakiyama et al 2015, Demirdas et al 2017, Micale et al 2019, Rymen et al 2019, Brisset et al 2020, Green et al 2020, Watanabe et al 2021]. The following description of the phenotypic features associated with this condition is based on these reports. It is important to note that the majority of affected individuals were diagnosed in adulthood.
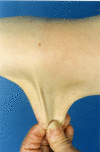
Skin. People with TNXB-related clEDS invariably have hyperextensible skin.
Although absence of atrophic scarring is one of the features that distinguishes TNXB-related clEDS from cEDS, mild atrophic scarring (not including cigarette paper scarring and hemosiderosis) has been reported in seven affected individuals.
Easy bruising is reported in the majority of affected individuals. In one individual a suspicion of nonaccidental injury had been raised due to excessive bruising.
Hematomas are frequently encountered.
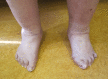
Musculoskeletal. Generalized joint hypermobility is always present in affected individuals, and many have recurrent joint (sub)luxations. Foot deformities (listed in Suggestive Findings) are present in the majority; hand abnormalities (acrogeric hands, mallet finger[s], clinodactyly, and brachydactyly) are less frequently reported [Demirdas et al 2017, Green et al 2020].
Significant variability between both unrelated and related affected individuals in the severity of musculoskeletal symptoms and their effect on day-to-day function has been reported.
The severity of symptoms in middle-aged individuals can range from joint hypermobility without complications to being wheelchair-bound due to severe and painful foot deformities, joint dislocations, and fatigue [
Green et al 2020].
Edema of the ankles and/or feet has been described in 14 of 56 individuals and could not be attributed to a cardiac etiology.
Cardiovascular
Vascular
fragility has been reported in 15 of 56 (27%) affected individuals, with three of 56 (5%) experiencing major medical events due to vascular fragility.
Ten individuals experienced frequent subconjunctival hemorrhages.
Rupture of a right brachial vein was reported in a woman age 26 years [
Micale et al 2019].
A man age 58 years had a thoraco-abdominal aortic aneurysm and aneurysm of both the common iliac artery and superior mesenteric artery [
Demirdas et al 2017].
An affected individual who died in his sixth decade due to a bowel rupture had aneurysmal abdominal arteries on postmortem examination [
Demirdas et al 2017].
One individual required surgery for two separate incidences of spontaneous compartment syndrome in his right and left arm at age 30 and 31 years, respectively [
Green et al 2020].
One individual developed a spontaneous left-calf hematoma that had to be drained surgically [
Green et al 2020].
This individual also developed a right-arm cephalic vein thrombosis and pulmonary embolism during admission for adrenal crisis.
She was subsequently started on anticoagulant therapy and shortly after required a hospital admission for spontaneous subcutaneous hematoma of the lower half of the body, causing acute anemia and requiring blood transfusion.
Valvular anomalies. Mild valvular abnormalities, often involving the mitral valve, have been reported in nine of 56 (16%) of affected individuals.
Cardiomyopathy was detected in three of 56 (5%) individuals (postpartum, dilated, and unspecified, respectively). It remains unclear if this represents rare co-occurrences of cardiomyopathy with TNXB-related clEDS or if cardiomyopathy is a rare feature of TNXB-related clEDS.
Neuromuscular
Subjective muscle weakness has been reported in about one third of affected individuals. Based partially on physiologic studies in affected individuals, a dose-effect relation of TNX levels and degree of neuromuscular involvement has been suggested [
Castori & Voermans 2014].
The most elaborate study included ten individuals with
TNXB-related clEDS among a group of 40 individuals with EDS of varying types [
Voermans et al 2009].
Those with TNXB-related clEDS generally had moderate neuromuscular complaints, mild reduced sensation, muscle weakness, and functional impairment on physical examination.
Moderate polyneuropathy and mild abnormal motor unit action potentials were seen on clinical neurophysiologic studies.
Muscle ultrasound demonstrated increased echo intensity.
Muscle biopsy showed mild myopathic changes in some affected individuals.
It has been hypothesized that neuropathy may be linked to increased vulnerability of peripheral nerves to stretching/pressure due to TNX deficiency [
Castori & Voermans 2014].
Atrophy of the muscles in the hands and feet has been reported in 4% of affected individuals, although this feature was not assessed by
Green et al [2020] in their cohort of 20 affected individuals. It is unclear whether this is a characteristic feature of
TNXB-related clEDS.
Gastrointestinal
Rupture. Nine affected individuals have been reported with gastrointestinal fragility leading to a total of 14 gastrointestinal events. Four of these individuals had more than one gastrointestinal event in different locations, and four had a gastrointestinal event during an invasive procedure. One of these latter four had three events, two of which occurred after an invasive procedure. Age at which first gastrointestinal complications occurred varied from 36 years to 59 years. These findings imply a degree of tissue fragility with complications resulting from invasive procedures. The nine affected individuals and their respective events were as follows:
A man age 36 years with a perforation of a colonic diverticulum, who developed multiple abscesses requiring partial colectomy, which was complicated by a second small-bowel perforation [
Lindor & Bristow 2005]
A man age 57 years who experienced an esophageal rupture possibly resulting from an ultrasound probe [
Hendriks et al 2012]
A woman age 45 years who had a diverticular perforation of the sigmoid colon and duodenal perforation after ileus tube insertion [
Sakiyama et al 2015]
An individual age 48 years who had a bowel perforation as a result of diverticulitis [
Demirdas et al 2017]
An individual age 38 years with a colonic perforation during a colonoscopy [
Brisset et al 2020]
A woman age 42 years who had a spontaneous perforation of the small bowel for which an intestinal specimen was reported to be "very fragile" [
Rymen et al 2019]
Spontaneous transverse colon perforation at age 51 years followed by a second perforation of the small bowel three days postoperative [
Green et al 2020]
Esophageal perforation at age 55 years during a gastroscopy, followed by a spontaneous small bowel perforation at age 56 years, and one small bowel perforation after a nasojejunal barium study at age 59 years [
Green et al 2020]
Note:
Demirdas et al [2017] reported death as a result of infection following bowel perforation of an individual in the sixth decade as unpublished data (not included in the cross-sectional analysis of 17 individuals represented in the publication).
Diverticular disease has been noted in ten of 56 affected individuals. It is hypothesized that affected individuals may be more prone to structural defects along the walls of the gastrointestinal tract, which can predispose to diffuse diverticulosis, diverticulitis, and resulting bowel perforation as well as to perforation during invasive procedures and spontaneous perforation [
Green et al 2020].
Gastrointestinal bleeding has been reported in three of 56 affected individuals. It was not specified whether this occurred spontaneously or due to another gastrointestinal issue.
Other tissue fragility. Tracheal rupture possibly due to intubation has been reported in a woman age 41 years [Besselink-Lobanova et al 2010]. A woman age 47 years had extensive surgical emphysema within the subcutaneous tissues of her face and computed tomography (CT) scans of the sinuses revealed a defect of the left nasal cartilages anteriorly, allowing air to track into the soft tissues. It was felt that vigorous nose blowing had most likely been the cause of the emphysema. This may also point to tissue fragility in individuals with TNXB-related clEDS [Green et al 2020].
Organ prolapse. As in other types of EDS, vaginal/uterus/rectal prolapse are more frequently encountered and have been reported in 21% of individuals with TNXB-related clEDS.