Clinical Characteristics
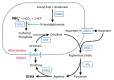
Severity of the urea cycle defect is influenced by the position of the defective protein in the pathway and the severity of the defect (see ).
Severe deficiency or total absence of activity of any of the first four enzymes in the pathway (CPS1, OTC, ASS1, and ASL) or the cofactor producer (NAGS) results in the accumulation of ammonia and other precursor metabolites during the first few days of life. Because no effective secondary clearance system for ammonia exists, complete disruption of this pathway results in the rapid accumulation of ammonia and development of related symptoms.
Presentation. Individuals with complete defects normally present in the newborn period, when the immaturity of the neonatal liver accentuates defects in the urea cycle enzymes [Pearson et al 2001, Summar 2001, Summar & Tuchman 2001].
Infants with a urea cycle disorder appear normal at birth but rapidly develop cerebral edema and the related signs of lethargy, anorexia, hyper- or hypoventilation, hypothermia, seizures, neurologic posturing, and coma.
Because newborns are usually discharged from the hospital within one to two days after birth, the symptoms of a urea cycle disorder often develop when the child is at home and may not be recognized in a timely manner by the family and primary care physician.
The typical initial symptoms of a child with hyperammonemia are nonspecific [Summar 2001, Kölker et al 2015]:
Symptoms progress from somnolence to lethargy and coma.
Abnormal posturing and encephalopathy are often related to the degree of central nervous system swelling and pressure on the brain stem [
Summar 2001].
About 50% of neonates with severe hyperammonemia may have seizures, some without overt clinical manifestations.
Individuals with closed cranial sutures are at higher risk for rapid neurologic deterioration from the cerebral edema that results from ammonia elevation.
Hyperventilation secondary to the effect of hyperammonemia on the brain stem, a common early finding in hyperammonemic attacks, results in respiratory alkalosis.
Hypoventilation and respiratory arrest follow as pressure increases on the brain stem.
In milder (or partial) urea cycle enzyme deficiencies, ammonia accumulation may be triggered at almost any time of life by illness or stress (e.g., surgery, prolonged fasting, holidays, the peripartum period), resulting in multiple mild elevations of plasma ammonia concentration.
Hyperammonemia in the milder defects is typically less severe and the symptoms more subtle than the neonatal presentation of a UCD.
In individuals with partial enzyme deficiencies, the first recognized clinical episode may be delayed for months or years.
Although the clinical abnormalities vary somewhat with the specific urea cycle disorder, in most the hyperammonemic episode is marked by loss of appetite, vomiting, lethargy, and behavioral abnormalities [
Gardeitchik et al 2012].
Sleep disorders, delusions, hallucinations, and psychosis may occur.
An encephalopathic (slow-wave) EEG pattern may be observed during hyperammonemia and nonspecific brain atrophy seen subsequently on MRI.
Defects in the final enzyme in the pathway (ARG1) cause hyperargininemia, a more subtle disorder involving neurologic symptoms; however, neonatal hyperammonemia has been reported (see Arginase Deficiency).
Defects in the two amino acid transporters (ORNT1 and citrin deficiency) may both cause hyperammonemia. However, ORNT1 deficiency may also present with chronic liver dysfunction. Citrin deficiency typically only presents with hyperammonemia in adolescence or adulthood, but may present in infants with neonatal intrahepatic cholestasis, and in older children with failure to thrive.
Neurologic aspects of UCDs. Ammonia can cause brain damage through a variety of proposed mechanisms, a major component of which is cerebral edema through increased glutamine. The specific roles of ammonia, glutamate, and glutamine in cerebral edema are still under investigation [Gropman et al 2007, Lichter-Konecki 2008, Lichter-Konecki et al 2008, Albrecht et al 2010, Braissant et al 2013].
Damage resulting from acute hyperammonemia in infancy resembles that seen in hypoxic-ischemic events or stroke. The most vulnerable areas are the insular cortex, which represents deep white matter. With prolonged hyperammonemia, the parietal, occipital, and frontal regions are affected. This is best appreciated on T2-weighted MRI sequences or on diffusion tensor imaging.
Neuroimaging may be helpful in identifying affected areas of the brain. However, MRI findings may lag behind clinical changes. In fact, early imaging may be normal as some degree of injury must occur before macroscopic changes are seen on MRI.
Chronic hyperammonemia may disrupt ion-gradients and neurotransmitters, transport of metabolites, mitochondrial function, and the alpha-ketoglutarate/ glutamate/glutamine ratio.
Seizures are common in acute hyperammonemia and may result from cerebral damage. Recent findings suggest that subclinical seizures are common in acute hyperammonemic episodes, especially in neonates, and their effects on cerebral metabolism in an otherwise compromised state should be addressed (see Management, Treatment of Acute Manifestations). These seizures may be seen during the rise of glutamine even before ammonia levels are maximal [Wiwattanadittakul et al 2018].
Survival and intellectual outcome. Historically the outcome of newborns with hyperammonemia was considered poor [Brusilow 1995]. With rapid identification and current treatment strategies, survival of neonates with hyperammonemia has improved dramatically in the last few decades. See Summar [2001], Summar & Tuchman [2001], Enns et al [2007], Summar et al [2008], Tuchman et al [2008], and Krivitzky et al [2009].
More recent data from the NIH-sponsored longitudinal study on patients treated with the more recent protocols show IQ measures within a less severe range as summarized in Table 1.
Table 1.
Cognitive and Adaptive Outcome in Children with UCD Age 3-16 Years
View in own window
| Age Group | Age 3-5 Years | Age 6-16 Years |
|---|
| Age at Onset | Neonatal 1
(n=5) | Late 2
(n=7) | Neonatal 1
(n=8) | Late 2
(n=39) |
|---|
|
Assessment
|
WASI/WPPSI-III 3
composite scores 4 (SD)
|
Verbal IQ
| 81.3 (16.6) | 101.7 (24.4) | 72.9 (14.3) | 94.3 (21.7) |
|
Performance IQ
| 77.7 (15.0) | 95.6 (17.4) | 74.4 (11.7) | 89.5 (20.4) |
|
Full scale IQ
| 77.7 (16.3) | 99.6 (22.6) | 71.4 (12.8) | 94.1 (22.0) |
|
ABAS-II 5 general adaptive composite 4 (SD)
| 73.2 (31.2) | 91.4 (23.6) | 66.0 (17.9) | 84.4 (21.6) |
- 1.
Clinical presentation in 1st month
- 2.
Clinical onset after 1st month or diagnosis based on family history
- 3.
Wechsler Abbreviated Scales of Intelligence / Wechsler Preschool and Primary Scale of Intelligence, 3rd Edition
- 4.
Clinically significant difference between groups for cognitive and adaptive outcome
- 5.
Adaptive Behavior Assessment System, 2nd Edition
While hyperammonemia is thought to be the main contributor to brain damage in UCDs, other factors, such as adverse effects on the nitric oxide production system [Nagamani et al 2012], may also contribute. For instance, neonates with CPS1 deficiency or OTC deficiency have more severe hyperammonemia than those with ASS1 deficiency or ASL deficiency; however, their intellectual outcomes appear similar [Ah Mew et al 2013].
In a recent study, asymptomatic female carriers of OTC deficiency demonstrated no significant differences in cognitive function compared to control participants until they were cognitively challenged with fine motor tasks, measures of executive function, and measures of cognitive flexibility [Sprouse et al 2014].