Clinical Description
Williams syndrome (WS) is a multisystem disorder characterized by cardiovascular disease (elastin arteriopathy, most often manifesting as supravalvar aortic stenosis), developmental delay, usually mild intellectual disability, a specific cognitive profile, unique personality characteristics, connective tissue abnormalities, growth abnormalities, other endocrine manifestations, and distinctive facies. Additional manifestations can include sleep problems, ocular issues, hearing loss, dental problems, gastrointestinal difficulties, urinary tract abnormalities, and musculoskeletal issues.
Table 2.
Williams Syndrome: Frequency of Select Features
View in own window
| System | Feature | % of Persons w/Feature | Comment |
|---|
|
Neurodevelopment
| Developmental delay | 100% | Average age for walking & talking is 21 mos. |
| Hypotonia | 80% | |
| Intellectual disability | 75% | Typically mild |
| Characteristic cognitive profile | 95% | Severe impairment of visuospatial construction, relative strength in language |
|
Neurobehavioral
| Unique personality | 95% | Overfriendliness, difficulty w/emotional regulation |
| Anxiety (non-social) | 80% | Specific phobia most common |
| ADHD | 65% | |
| Sleep disorders | 65% | |
| Autism spectrum disorder | 10%-20% | |
|
Cardiovascular
| Any arterial stenosis | 80% | Elastin arteriopathy |
| SVAS | 75% | |
| PPS | 40%-60% | Typically improves w/time |
| Hypertension | 50% | Renal artery stenosis may contribute |
| Mitral valve prolapse | 15% | |
| QTc prolongation | 13% | |
|
Ocular/visual
| Esotropia | 50% | |
| Hyperopia | 60% | |
|
Auditory
| Recurrent otitis media | 50% | |
| Hypersensitivity to sound | 90% | |
| Sensorineural hearing loss | >60% | Present in >90% of adults |
|
Dental
| Microdontia | 95% | |
| Malocclusion | 85% | |
|
Gastrointestinal
| Feeding difficulties | 70% | |
| Constipation | 50% | |
| Umbilical hernia | 50% | |
| Inguinal hernia | 40% | |
| Colon diverticula | 30% | |
| Rectal prolapse | 15% | |
|
Urinary tract
| Urinary frequency | 70% | |
| Enuresis | 50% | |
| Bladder diverticula | 50% | |
|
Musculoskeletal
| Joint hypermobility | 90% | Most common in young children |
| Joint contractures | 50% | |
| Scoliosis | 20%-35% | |
Integument /
connective tissue
| Hoarse voice | 90% | |
| Soft, lax skin | 90% | |
|
Endocrine
| Short stature | 50% | Mean adult height is <3rd centile |
| Early puberty | 50% | |
| Hypercalcemia | 20%-40% | Clinically significant hypercalcemia typically occurs at age <2 yrs |
| Hypercalciuria | 30% | Nephrocalcinosis in 5% |
| Hypothyroidism | 10% | Subclinical in 30% |
| Diabetes mellitus | 20% | More common in adults |
|
Craniofacial
| Characteristic facial features | 100% | See , , , and . |
| Microcephaly | 30% | |
ADHD = attention-deficit/hyperactivity disorder; PPS = peripheral pulmonic stenosis; SVAS = supravalvar aortic stenosis
Infancy. Infants with WS are often born post-term, and 50% of infants are small for gestational age [Li et al 2022]. Feeding difficulties leading to poor weight gain are common, including gastroesophageal reflux, disordered suck and swallow, textural aversion, and vomiting. Prolonged colic (lasting longer than four months) may be related to gastroesophageal reflux, chronic constipation, and/or idiopathic hypercalcemia.
Developmental delay. Infants with WS are hypotonic and typically have hyperextensible joints, resulting in delayed attainment of motor milestones [Morris & Mervis 2021]. Walking usually occurs by age 24 months. Speech is also delayed but later becomes a relative strength [Kozel et al 2021]. Fine motor difficulties are present at all ages.
Cognitive abilities. Intellectual disability, usually mild, occurs in 75% of individuals with WS. The cognitive profile is distinctive, consisting of strengths in verbal short-term memory and language but extreme weakness in visuospatial constructive cognition. As a result, children with WS usually score higher on verbal subtests than on tests measuring visuospatial construction [Mervis & Greiner de Magalhães 2022]. No difference in IQ between males and females is reported, and IQ is stable throughout childhood [Mervis et al 2012].
Academically, individuals with WS perform relatively well in reading, and adults may read at a high school level, though the range of achievement is wide. Reading skills correlate with method of reading instruction rather than IQ, with the highest reading skills following systemic phonics instruction [Mervis & Greiner de Magalhães 2022]. Difficulty with writing, drawing, and mathematics is significant, although many adults with WS are able to perform simple addition.
Unique personality/behavior. The characteristic personality profile of WS includes overfriendliness, social disinhibition, excessive empathy, attention problems, and non-social anxiety. Other common behavior problems include difficulty with sensory modulation/processing, emotional regulation, and perseveration. Some individuals have overlapping symptoms with autism spectrum disorder, such as restricted interests and repetitive behavior. Children with WS who meet diagnostic criteria for autism spectrum disorder (ASD; 10%-20%) fit in the active-but-odd subtype rather than the aloof subtype of ASD [Klein-Tasman et al 2018]. In children, attention-deficit/hyperactivity disorder occurs in 65% of individuals and anxiety disorder in 57% (usually specific phobias) [Leyfer et al 2006]. Anxiety is common across the life span; longitudinal studies of anxiety indicate a prevalence of 80% [Woodruff-Borden et al 2010]. Pharmacotherapy for psychiatric disorders in WS should take into account the medical comorbidities associated with WS [Thom et al 2021]. For instance, use of stimulants (which can increase blood pressure and heart rate) to treat ADHD should be discussed with the individual's cardiologist; atomoxetine is a non-stimulant alternative [Thom et al 2021].
Adaptive behavior is less than expected for IQ in both children and adults [Howlin & Udwin 2006, Mervis & Pitts 2015], and adversely affects the ability of adults with WS to function independently (less than 10% live independently). There is typically a relative strength in socialization and communication skills, with significant weakness in motor skills and daily living skills. Executive function deficits occur in 70% of children and adolescents with WS. Difficulties with behavioral and emotional regulation are associated with behavior issues and poor adaptive behavior; difficulty with inhibition is associated with attention problems; and difficulty with flexibility in shifting focus is associated with anxiety [Greiner de Magalhães et al 2022].
Sleep disorders occur in 65% of affected individuals and include increased sleep latency and decreased sleep efficiency [Goldman et al 2009, Mason et al 2011]. Abnormal or absent nocturnal melatonin peak has been documented [Sniecinska-Cooper et al 2015, Santoro et al 2016]; melatonin treatment may be helpful [Martens et al 2017, Morris & Mervis 2021]. Sleep-related breathing disorder (reported in 15%) and excessive daytime sleepiness (reported in 30%) were associated with more externalizing behavior problems in toddlers with WS at age two years [Greiner de Magalhães et al 2020].
Cardiovascular disease occurs in 80% of individuals with WS [Collins 2018, Honjo et al 2022]. Elastin arteriopathy is the major cause of morbidity and mortality. Any artery may be narrowed, but supravalvar aortic stenosis (SVAS) is most common and may worsen over time, especially in the first five years of life [Collins et al 2010b]. SVAS can be either a discrete hourglass stenosis or diffuse aortic stenosis. If untreated, the resultant increase in arterial resistance leads to elevated left heart pressure, cardiac hypertrophy, and cardiac failure. Progression is more likely if the stenosis is moderate or severe and presents in infancy or early childhood. Mild SVAS is less likely to progress. Approximately one third of children with SVAS will require surgical correction.
Peripheral pulmonic stenosis (PPS) is common in infancy but usually improves over time without intervention. However, individuals with combined SVAS and PPS (biventricular outflow tract obstruction) may develop biventricular hypertrophy and hypertension, increasing the risk for myocardial ischemia, dysrhythmias, and sudden death [Pham et al 2009]. The overall 30-year survival rate for children undergoing interventions for obstructive lesions is 90% in North America [Zinyandu et al 2023]. Individuals with either isolated left heart obstructive lesions or isolated right heart obstructive lesions have better survival rates than those with combined disease (96% vs 83%). In-hospital mortality was 2.5%, and 9% required reoperation (mostly for recurrent SVAS) [Zinyandu et al 2023].
Coronary artery stenosis has been implicated as a cause of sudden death in individuals with WS [Bird et al 1996]. Coronary artery stenosis or dilatation are found in 10%-30% of individuals with WS, and coronary blood flow may be restricted at the sinuses of Valsalva in the setting of narrowed sinotubular junction [Brown et al 2018]. The incidence of sudden death in one cohort of 293 individuals with WS was one in 1,000 patient years, which is 25 to 100 times higher than the age-matched population [Wessel et al 2004].
The prevalence of hypertension in individuals with WS is 40%-50%. Hypertension may present at any age [Bouchireb et al 2010] and may be secondary to renal artery stenosis in some instances [Honjo et al 2022]. Increased vascular stiffness has been documented in individuals with WS and responds to antihypertensive medication [Kozel et al 2014].
Mitral valve prolapse and aortic insufficiency have been reported in adults [Morris et al 1990, Collins et al 2010a].
Prolonged QTc has been reported in 13.6% of individuals with WS; screening for repolarization abnormalities is recommended [Collins et al 2010a, Brink et al 2022].
Anesthesia and sedation are associated with an increased risk for adverse events, including cardiac arrest, in individuals with WS [Olsen et al 2014]. Sedation and anesthesia risk assessment and management guidelines have been developed [Schmidt et al 2021, Staudt & Eagle 2021] with the goal of maintaining intravascular volume and a stable blood pressure to optimize coronary perfusion [Schmidt et al 2021]. In centers using these strategies specifically for individuals with WS, morbidity and mortality is decreased. Brown et al [2018] reported no adverse events in 90% of individuals undergoing cardiac procedures, and no adverse events in 95% of individuals undergoing noncardiac procedures. Schmidt et al [2021] compared a historical group with an intervention group after new anesthesia guidelines were adopted for WS and found that incidence of adverse cardiac events in the intervention group was 2% compared to 6% in the historical group.
Stenosis of additional arteries has been reported. Neurovascular abnormalities are rarely reported but may result in stroke [Cherniske et al 2004]. Stenosis of the mesenteric arteries may contribute to abdominal pain.
Ocular manifestations. Lacrimal duct obstruction, hyperopia (67%), and esotropia (50%) are common in individuals with WS [Weber et al 2014]. Cataracts have been reported in adults [Cherniske et al 2004].
Ears and hearing. Chronic otitis media is seen in 50% of affected individuals. Increased sensitivity to sound is common (90%), and individuals with WS report discomfort at 20 decibels lower than controls [Gothelf et al 2006]. Many affected individuals report specific phobias for certain sounds [Levitin et al 2005]. Hyperacusis occurs in 35% and is associated with absence of contralateral acoustic reflexes [Silva et al 2021].
Progressive sensorineural hearing loss has been observed; mild-to-moderate hearing loss is detected in 63% of children and 92% of adults [Gothelf et al 2006, Marler et al 2010]. Mild-to-moderate high-frequency sensorineural hearing loss is common in adults, as is excessive buildup of ear wax [Cherniske et al 2004].
Dental findings include generalized diastemas (70%), hypodontia (50%), microdontia, enamel hypoplasia, and malocclusion (angle class II and III) [Castro et al 2019].
Gastrointestinal manifestations. Individuals with WS have sensory defensiveness; difficulty with food textures leads to problems in transitioning from breast milk or formula to solid foods in infancy.
Chronic abdominal pain is common in children and adults with WS; possible causes include gastroesophageal reflux, hiatal hernia, peptic ulcer disease, cholelithiasis, diverticulitis, ischemic bowel disease, chronic constipation, and somatization of anxiety. The prevalence of diverticulitis is increased in adolescents [Stagi et al 2010] and adults with WS [Partsch et al 2005]. Complications of constipation may include rectal prolapse, hemorrhoids, or intestinal perforation.
Hypercalcemia may contribute to irritability, vomiting, constipation, and muscle cramps. Hypercalcemia is more common in infancy but may recur in adults [Morris et al 1990, Pober et al 1993].
Urinary tract abnormalities. Urinary frequency and enuresis are common in children with WS. Renal artery stenosis is found in 50% of individuals with WS, structural abnormalities of the urinary tract in 10%, bladder diverticulae in 50%, and nephrocalcinosis in fewer than 5% [Pober et al 1993, Pankau et al 1996, Sforzini et al 2002, Sammour et al 2006, Sammour et al 2014]. Bladder capacity is reduced, and detrusor overactivity is observed in 60% of affected individuals [Sammour et al 2006]. Average age of daytime urinary continence is four years; nocturnal continence occurs in 50% by age ten years. Nocturnal enuresis occurs in an estimated 3% of adults [von Gontard et al 2016].
Musculoskeletal and additional neurologic manifestations. The hypotonia and lax joints of the young child lead to abnormal compensatory postures to achieve stability. Older children and adults with WS typically have hypertonia and hyperactive deep-tendon reflexes. Gradual tightening of the heel cords and hamstrings occurs, resulting in a stiff and awkward gait, kyphosis, and lordosis by adolescence [Morris et al 1988, Kaplan et al 1989, Copes et al 2016]. Scoliosis is common [Damasceno et al 2014]. Ten percent of individuals with WS have radioulnar synostosis [Morris & Carey 1990]. Fine motor function is impaired, leading to difficulty with tool use and handwriting at all ages. Cerebellar signs in adults include ataxia, dysmetria, and tremor [Pober & Morris 2007].
Neuroimaging. Reduced brain size, reduced gray matter volume (especially in the parietal and occipital regions), and increased gyral complexity are seen on brain MRI [Jackowski et al 2009, Eisenberg et al 2010]. Reduced posterior fossa size coupled with preserved cerebellar size may contribute to Chiari I malformation found in some affected individuals [Pober & Filiano 1995, Mercuri et al 1997].
Integument and additional connective tissue manifestations. Most individuals have a hoarse or low-pitched voice; vocal cord abnormalities secondary to elastin deficiency are likely causative [Vaux et al 2003]. Soft, lax skin is typical. The hair grays prematurely [Morris et al 1988].
Growth deficiency. Poor weight gain is observed in 70% of infants. The growth pattern is characterized by prenatal growth deficiency, poor weight gain, and poor linear growth in the first four years, a rate of linear growth that is 75% of normal in childhood, and a brief pubertal growth spurt. Individuals with WS are shorter than predicted by midparental height [Levy-Shraga et al 2018]. The mean adult height is below the third centile. Specific growth curves for WS are available [Saul et al 1988, Martin et al 2007, Morris et al 2020]. For a systematic review of growth studies in WS cohorts, see de Sousa Lima Strafacci et al [2020]. Obesity or overweight is seen in 50% of older children and adults [Cherniske et al 2004, Stanley et al 2021]. A lipedema phenotype of the lower extremities in seen in 25% of adults [Shaikh et al 2018].
Puberty may occur early, and central precocious puberty is present in 18% of individuals with WS [Partsch et al 2002, Kim et al 2016]. Hormonal suppression with gonadotropin-releasing hormone agonist is well tolerated by girls with either early or precocious puberty, and treated girls are taller than WS controls [Spielmann et al 2015].
Hypercalcemia is most often symptomatic (irritability, vomiting, constipation) in the first two years of life [Martin et al 1984, Morris et al 1988, Kim et al 2016]. Hypercalcemia is associated with dehydration, hypercalciuria, and nephrocalcinosis. Compared to controls, higher median serum calcium levels are found in all age groups [Sindhar et al 2016]. The etiology of hypercalcemia in WS is unknown [Stagi et al 2016].
Additional endocrine problems. Endocrine abnormalities include hypothyroidism, including subclinical hypothyroidism (thyroid-stimulating hormone elevation with normal T3/T4 levels) [Palacios-Verdú et al 2015]. Prevalence of impaired glucose tolerance is 42%, and incidence of diabetes mellitus is increased in adolescents and adults with WS [Stanley et al 2021]. Low bone mineral density has been reported in 50% of adults with WS [Cherniske et al 2004] and is associated with low serum phosphate [Palmieri et al 2019].
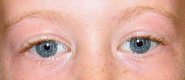
Distinctive facial features including broad forehead, bitemporal narrowing, periorbital fullness, a stellate/lacy iris pattern (see ), strabismus, short nose, broad nasal tip, malar flattening, long philtrum, thick vermilion of the upper and lower lips, wide mouth, malocclusion, small jaw, and large ear lobes are observed at all ages (see ). Young children have epicanthal folds, full cheeks, and small, widely spaced teeth (see ), while adults typically have a long face and neck, accentuated by sloping shoulders (see ).