Clinical Description
FLNA deficiency is prenatally or neonatally lethal in most males; therefore, the majority of affected individuals are female. To date, more than 100 individuals (both males and females) have been identified with loss-of-function variants in FLNA. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
FLNA Deficiency: Frequency of Select Features (Males and Females)
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
| Seizure disorder | 75%-90% | |
| Cardiovascular | 65% | Patent ductus arteriosus; dilatation & rupture of thoracic aorta; atrial & ventricular septal defects; valvular dystrophy; vasculopathy &/or coagulopathy → stroke |
| Pulmonary disease | 25% | Pulmonary hypertension, alveolar hypoplasia, emphysema, asthma, chronic bronchitis |
| ↓ gastric motility | 6% | Chronic intestinal pseudo-obstruction, feeding difficulties |
| Joint hypermobility | <15% | |
| Distally shortened digits | <5% | |
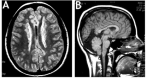
Seizure disorder. Approximately 88% of individuals (including males and females) with FLNA-related periventricular nodular heterotopia (PVNH) have presented with a seizure disorder [Guerrini & Carrozzo 2001]; in other series that included investigation of asymptomatic heterozygous mothers, the percentage was 63.3% [Lange et al 2015]. Many individuals are ascertained by MRI scan following a first seizure. As individuals are increasingly identified based on non-neurologic findings, a lower proportion of individuals with seizures will likely be confirmed. Age of seizure onset may be as early as the first years of life, but typically individuals present during childhood or adolescence, or even as adults, with an overall average age of seizure onset in the mid-teens [Fox et al 1998, Sheen et al 2001, Lange et al 2015]. The severity of the seizure disorder may range from mild (i.e., rare events, not requiring anti-seizure medication) to intractable seizures.
No correlation exists between the extent and severity of the nodular heterotopia seen radiographically and the clinical manifestations, though early seizure onset correlates with poorer developmental outcome [Lange et al 2015]. The ectopic heterotopias in some individuals act as foci for abnormal neuronal activity. Anatomic studies have shown aberrant projections extending from the periventricular heterotopias. Depth electrode recordings have demonstrated epileptogenic discharges from these nodules [Kothare et al 1998]. Thus, the seizure disorder appears to arise from the heterotopias in most individuals.
Cardiovascular findings. In a large cohort of 114 individuals from 80 families with loss-of-function FLNA variants and PVNH, 64.9% had a cardiovascular anomaly. Two thirds were structural cardiac defects, with patent ductus arteriosus, ventricular septal defects, and valve abnormalities being the most common [Chen et al 2018].
Almost 20% of individuals reported by Chen et al [2018] had thoracic aortic dilatation or aneurysm (TAA). Individuals with TAA typically had involvement of the aortic root and ascending aorta, and half of individuals with TAA had a sinus of Valsalva aneurysm [Feng et al 2006].
Isolated X-linked cardiac valvular dysplasia (XCVD) has been identified in individuals with FLNA missense variants associated with some retained FLNA function, typically resulting in myxomatous valvular dystrophy most commonly involving the mitral valve [Kyndt et al 2007, Le Tourneau et al 2018] although other valvular involvement was also noted. XCVD may necessitate cardiac surgery in some individuals. This phenotype was identified in individuals with one of three FLNA missense variants: p.Pro637Gln, p.His743Pro, or p.Gly288Arg. Affected males in the cohort had more severe valvular disease than females, who were sometimes unaffected [Le Tourneau et al 2018].
Pulmonary findings. Individuals with FLNA deficiency may develop varying degrees of pulmonary hypertension, which when severe has necessitated treatment with sildenafil. Several reported individuals (both male and female) had cardiorespiratory failure before age one year. The severity of the poorly defined respiratory disease, resembling bronchopulmonary dysplasia, has led to lung transplantation in several of these young children [Masurel-Paulet et al 2011, Clapham et al 2012, Lord et al 2014, Burrage et al 2017, Kremer et al 2019, Sasaki et al 2019].
Gastrointestinal manifestations. The spectrum of disease severity has ranged from constipation to chronic intestinal pseudo-obstruction (CIPO). Several individuals (primarily males), have been reported with delayed gastric motility [Negri et al 2020]. Clinical presentation typically includes feeding difficulties at birth, constipation, progressive weight loss, and congenital short bowel seen in isolation or in combination with intestinal malrotation [Oegema et al 2013]. Feeding issues may be present and improve with time, resolving after the first year of life. Conservative treatments including diet management were successful in some symptomatic older individuals [Oegema et al 2013]. Prolonged nutrition supplementation, gastrotomy, and/or jejunostomy tube feeding may be necessary in some individuals.
Males with FLNA loss-of-function variants have presented with severe abdominal wall defects, resulting in prune belly syndrome; characterized by: wrinkled, lax abdominal walls with partial or complete skeletal muscle deficiency; urinary tract dilatation with poorly contractile smooth muscle; and undescended testes [Iqbal et al 2020].
Isolated gastrointestinal manifestations. Loss-of-function FLNA variants have also been described as a cause of congenital short bowel syndrome in males and females. Males with X-linked congenital intestinal pseudo-obstruction (CIPO) have been found to have FLNA loss-of-function variants [van der Werf et al 2015, Negri et al 2020, Wade et al 2020]. CIPO includes reduced intestinal motility and a short, malrotated, and dilated small intestine. Severe intestinal dysmotility resulted in the need for long-term parenteral nutrition in several individuals [Jenkins et al 2018].
Joint hypermobility. In a case series of 47 individuals (2 males, 45 females) with FLNA-related PVNH, 14.7% presented with joint laxity. Joint dislocation can also occur, involving the hips and other joints [Lange et al 2015].
Skin abnormalities. Cutis laxa, hyperextensibility, and thin, soft, or translucent skin [Reinstein et al 2013, Ritelli et al 2017] may also be present, occurring independently or in combination.
Shortened digits. Some individuals are reported to have hypoplasia of the distal phalanges in general or of the great toe, or short proximal thumbs.
Macrothrombocytopenia. Individuals with FLNA deficiency can present with large platelets that may also be reduced in number, resulting in excessive bruising, bleeding, and slow wound healing [Nurden et al 2011]. Macrothombocytopenia can occur with missense, deletion, exon-skipping [Oda et al 2016], or truncation variants of FLNA. Isolated macrothrombocytopenia was reported in an individual with thrombocytopenia, bleeding, and an FLNA missense variant (p.Glu1795Lys) without other features of FLNA deficiency [Nurden et al 2011].
Cognitive function / dyslexia. Intelligence is normal to borderline. Formal cognitive testing of ten affected individuals (2 males, 8 females) with FLNA loss-of-function variants demonstrated an average IQ of 95, but also a strikingly high number of affected individuals with dyslexia [Chang et al 2005, Chang et al 2007, Reinstein et al 2012]. However, these results likely reflect a selection bias towards more severely affected individuals, since those with normal cognitive function usually would not have undergone a screening brain MRI or molecular testing.
Pregnancy complications. Women with FLNA deficiency may have an increased incidence of pregnancy loss as a result of spontaneous abortion of affected male pregnancies.
Other
Mosaicism. Somatic mosaicism for an A>G substitution at the intron 11 acceptor splice site was reported by Parrini et al [2004] in a male with bilateral PVNH. Sequence analysis and denaturing high-performance liquid chromatography of genomic DNA on a pool of hair roots, single hair roots, and white blood cells revealed that only 42% and 69% of the samples for hair and blood, respectively, had the pathogenic variant. Moreover, the affected male's daughter did not inherit the pathogenic variant, thought to be causal for the male phenotype. Other somatic pathogenic variants were subsequently reported [Jamuar et al 2014, González-Morón et al 2017].