Clinical Description
Affected individuals usually develop nystagmus within the first six months of life; the mean age of onset is two months. Nystagmus can be gaze-dependent oscillations or time-dependent oscillations (periodic alternating nystagmus) [Thomas et al 2008, Thomas et al 2011].
Nystagmus waveform characteristics are established by eye movement recordings that assess the following (see and ):
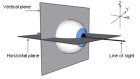
Eye rotation can occur about three axes (X, Y, Z). Torsional eye movements occur along the line of sight (X); horizontal and vertical eye movements occur along the Z and Y axes, respectively. The oscillations seen in FIN occur only in the horizontal plane. (more...)
The horizontal eye movement recordings in an individual with FIN (a) Gaze-dependent nystagmus. Note the right-beating pattern on right gaze.
Conjugacy
Plane of oscillations (horizontal, vertical, and torsional)
Pattern of oscillations (pendular, jerk, or bidirectional waveforms)
Direction of oscillations (quick phase)
Quantitative features of the waveform, including:
Frequency
Amplitude
Foveation dynamics (foveation is the period during which the eyes remain relatively still and the image is incident on the fovea)
Null point width (range of eye eccentricities in which the nystagmus is quietest)
Note: The quantitative features of the waveform can only be evaluated using eye movement recordings.
The above measurements also help in assessing the clinical severity of the nystagmus.
Conventionally, intensity (product of amplitude and frequency) is measured in order to describe the severity of nystagmus; however, foveation correlates best with visual function scores.
Foveation takes into account both the retinal image velocity and position of the image in relation to the fovea. An example of the measure of foveation is the NAFX (extended nystagmus acuity function), which assesses the standard deviation of the aforementioned parameters and the duration of the foveation.
Measuring intensity, foveation characteristics, and null point width before and after treatment provides an objective measure of the therapeutic response.
Numerous studies have shown that the predominant waveform changes with age (see Table 2). In a unique case report, eye movements were described and recorded before the onset of nystagmus [Gottlob 1997].
At the onset, large-amplitude, low-frequency horizontal eye movements (described as triangular eye movements) are seen. This waveform pattern is followed by a smaller-amplitude pendular or jerk waveform and development of foveation. Another study reported that the predominant waveform during the first six months was asymmetric pendular and jerk with extended foveation [Hertle et al 2002].
Table 2.
How the Infantile Nystagmus Waveform Evolves: An Example
View in own window
| Age | Waveform Description |
|---|
| 5 weeks 1 | No nystagmus |
| 7 weeks | Square wave jerk |
| 8 weeks | Small pendular nystagmus |
| 10 weeks | Large jerk type nystagmus |
| 14 weeks | Small pendular nystagmus |
| 7-12 months | Conjugate pendular nystagmus |
- 1.
The infant was initially part of another study looking at normal visual development.
In adults, a pendular waveform is more commonly associated with FRMD7-related infantile nystagmus (FIN) than with non-FRMD7 idiopathic infantile nystagmus (IIN) (see Differential Diagnosis) [Thomas et al 2008]. These oscillations are accentuated by attention, anxiety, attempts to fixate on an object, and directing the gaze away from the null zone.
Individuals with FIN report good visual acuity (typically >6/12) because the nystagmus waveform is interrupted by a foveation period and, in contrast to other forms of infantile nystagmus, FIN is not the result of sensory abnormalities (e.g., reduced visual acuity resulting from foveal hypoplasia) (see Differential Diagnosis).
An abnormal head posture is seen in approximately 15% of affected individuals. Affected individuals may assume an anomalous head posture if they have an eccentric null zone. Titubation of the head is observed in some individuals. However, affected individuals do not report any tremor of the limbs or trunk or any balance or coordination issues.
Oscillopsia, the illusion of movement in one's surroundings, is very rarely reported in FIN. This may result in part from the presence of foveation periods during the waveform. However, an affected individual may complain of oscillopsia when looking at a position of gaze in which the nystagmus is more pronounced or when the individual is tired.
Affected females report slightly better visual acuity than affected males. However, no notable differences in amplitude, frequency, and waveform of nystagmus are observed between males and females.
The optokinetic response (OKR) is abnormal, with either low gains or reversal patterns described [Thomas et al 2008]. In individuals with periodic alternating nystagmus and a FRMD7 pathogenic variant no OKR is observed [Thomas et al 2008, Thomas et al 2011]. In unaffected female carriers a subnormal OKR has been described [Thomas et al 2008].
Optical coherence tomography studies have shown either normal fovea morphology or grade 1 foveal hypoplasia [Thomas et al 2014]. Similarly, optic nerve morphology can be abnormal in FIN [Thomas et al 2014, Choi et al 2018].