

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
McEntyre J, Ostell J, editors. The NCBI Handbook [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2002-.

This publication is provided for historical reference only and the information may be out of date.

Summary
Entrez is the text-based search and retrieval system used at NCBI for all of the major databases, including PubMed, Nucleotide and Protein Sequences, Protein Structures, Complete Genomes, Taxonomy, OMIM, and many others. Entrez is at once an indexing and retrieval system, a collection of data from many sources, and an organizing principle for biomedical information. These general concepts are the focus of this chapter. Other chapters cover the details of a specific Entrez database (e.g., PubMed in Chapter 2) or a specific source of data (e.g., GenBank in Chapter 1).
Entrez Design Principles
History
The first version of Entrez was distributed by NCBI in 1991 on CD-ROM. At that time, it consisted of nucleotide sequences from GenBank and PDB; protein sequences from translated GenBank, PIR, SWISS-PROT, PDB, and PRF; and associated citations and abstracts from MEDLINE (now PubMed and referred to as PubMed below). We will use this first design to illustrate the principles behind Entrez.
Entrez Nodes Represent Data
An Entrez “node” is a collection of data that is grouped together and indexed together. It is usually referred to as an Entrez database. In the first version of Entrez, there were three nodes: published articles, nucleotide sequences, and protein sequences. Each node represents specific data objects of the same type, e.g., protein sequences, which are each given a unique ID (UID) within that logical Entrez Proteins node. Records in a node may come from a single source (e.g., all published articles are from PubMed) or many sources (e.g., proteins are from translated GenBank sequences, SWISS-PROT, or PIR) (Figure 1).
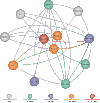
Figure 1
The original version of Entrez had just 3 nodes: nucleotides, proteins, and PubMed abstracts. Entrez has now grown to nearly 20 nodes.
Note that the UID identifies a single, well-defined object (i.e., a particular protein sequence or PubMed citation). There may be other information about objects in nodes, such as protein names or EC numbers, that may be used as index terms to find the record, but these pieces of information are not the central organizing principle of the node. Each data object represents a stable, objective observation of data as much as possible, rather than interpretations of the data, which are subject to change or confusion over time or across disciplines. For example, barring experimental error, a particular mRNA sequence report is not likely to change over the years; however, the given name, position on the chromosome, or function of the protein product may well change as our knowledge develops. Even a published article is a stable observation. The fact that the article was published at a certain time and contained certain words will not change over time, although the importance of the article topic may change many times.
Entrez Nodes Are Intended for Linking
Another criterion for selecting a particular data type to be an Entrez node is to enable linking to other Entrez nodes in a useful and reliable way. For example, given a protein sequence, it is very useful to quickly find the nucleotide sequence that encodes it. Or given a research article, it is useful to find the sequences it describes, if any.
Links between Nodes
One way to achieve this is to put all of the information into one record. For example, many GenBank records contain pertinent article citations. However, PubMed also contains the article abstract and additional index terms (e.g., MeSH terms); furthermore, the bibliographic information is also more carefully curated than the citation in a GenBank entry. It therefore makes much more sense to search for articles in PubMed rather than in GenBank.
When a subset of articles has been retrieved from PubMed, it may be useful to link to sequence information associated with the abstracts. The article citation in the GenBank record can be used to establish the link to PubMed and, conversely, to make the reciprocal link from the PubMed article back to the GenBank record. Treating each Entrez node separately but enabling linking between related data in different nodes means that the retrieval characteristics for each node can be optimized for the characteristics and strengths of that node, whereas related data can be reached in nodes with different strengths.
This approach also means that new connections between data can be made. In the example above, the GenBank record cited the published article, but there was no link from that article in PubMed to the sequence until Entrez made the reciprocal link from PubMed. Now, when searching articles in PubMed, it is possible to find this sequence, although no PubMed records have been changed. Because of this design principle, the Entrez system is richly interconnected, although any particular association may originate from only one record in one node.
Links within Nodes
Another type of linking in Entrez is between records of the same type, often called “neighbors”, in sequence and structure nodes. Most often these associations are computed at NCBI. For example, in Entrez Proteins, all of the protein sequences are “BLASTed” against each other, and the highest-scoring hits are stored as indexes within the node. This means that each protein record has associated with it a list of highly similar sequences, or neighbors.
Again, associations that may not be present in the original records can be made. For example, a well-annotated SWISS-PROT record for a particular protein may have fields that describe other protein or GenBank records from which it was derived. At a later date, a closely related protein may appear in GenBank that will not be referenced by the SWISS-PROT record. However, if a scientist finds an article in PubMed that has a link to the new GenBank record, that person can look at the protein and then use the BLAST-computed neighbors to find the SWISS-PROT record (as well as many others), although neither the SWISS-PROT record nor the new GenBank record refers to each other anywhere.
Entrez Nodes Are Intended for Computation
There are many advantages to establishing new associations by computational methods (as in the GenBank–SWISS-PROT example above), especially for large, rapidly changing data sets such as those in biomedicine.
As computers get faster and cheaper, this type of association can be made more efficiently. As data sets get bigger, the problem remains tractable or may even improve because of better statistics. If a new algorithm or approach is found to be an improvement, it is possible to apply it over the whole data set within a practical timescale and by using a reasonable number of resources. Any associations that require human curation, such as the application of controlled vocabularies, do not scale well with rapidly growing sets of data or evolving data interpretations. Although these manual kinds of approaches certainly add value, computational approaches can often produce good results more objectively and efficiently.
Entrez Is a Discovery System
A data-retrieval system succeeds when you can retrieve the same data you put in. A discovery system is intended to let you find more information than appears in the original data. By making links between selected nodes and making computed associations within the same node, Entrez is designed to infer relationships between different data that may suggest future experiments or assist in interpretation of the available information, although it may come from different sources.
The ability to compare genotype information across a huge range of organisms is a powerful tool for molecular biologists. For example, this technique was used in the discovery of a gene associated with hereditary nonpolyposis colon cancer (HNPCC). The tumor cells from most familial cases of HNPCC had altered, short, repeated DNA sequences, suggesting that DNA replication errors had occurred during tumor development. This information caused a group of investigators to look for human homologs of the well-characterized Escherichia coli DNA mismatch repair enzyme, MutS (1). Mutants in a MutS homolog in yeast, MSH2, showed expansion and contraction of dinucleotide repeats similar to the mutation found in the human tumor cells. By comparing the protein sequences between the yeast MSH2, the E. coli MutS, and a human gene product isolated and cloned from HNPCC colorectal tumor, the researchers could show that the amino acid sequences of all three proteins were very similar. From this, they inferred that the human gene, which they called hMSH2, may also play a role in repairing DNA, and that the mutation found in tumors negatively affects this function, leading to tumor development.
The researchers could connect the functional data about the yeast and bacterial genes with the genetic mapping and clinical phenotype information in humans. Entrez is designed to support this kind of process when the underlying data are available electronically. In PubMed, the research paper about the discovery of hMSH2 (1) has links to the protein sequence, which in turn has links to “neighbors” (related sequences). There are lots of records for this protein and its relatives in many organisms, but among them are the proteins from yeast and E. coli that prompted the study. From those records there are links back to the PubMed abstracts of articles that reported these proteins. PubMed also has a “neighbor” function, Related articles, that represents other articles that contain words and phrases in common with the current record. Because phrases such as “Escherichia coli”, “mismatch repair”, and "MutS" all occur in the current article, many of the articles most related to this one describe studies on the E. coli mismatch repair system. These articles may not be directly linked to any sequence themselves and may not contain the words “human” or “colon cancer” but are relevant to HNPCC nonetheless, because of what the bacterial system may tell us.
Entrez Is Growing
The original three-node Entrez system has evolved over the past 10 years to include more nodes (Figure 1). These include:
- 1.
Taxonomy, which is organized around the names and phylogenetic relationships of organisms
- 2.
Structure, organized around the three-dimensional structures of proteins and nucleic acids
- 3.
Genomes, in which each record represents a chromosome of an organism
- 4.
Online Mendelian Inheritance in Man (OMIM), a text-based resource organized around human genes and their phenotypes
- 5.
PopSet, consisting of collections of aligned sequences from a single population study
- 6.
Books, representing published books in biomedicine
More nodes are planned for addition in the near future. Each one of these nodes is richly connected to others. Each offers unique information and unique new relationships among its members. The combination of new links and new relationships increases the chances for discovery. The addition of each new node creates different paths through the data that may lead to new connections, without more work on the old nodes.
How Entrez Works
Entrez integrates data from a large number of sources, formats, and databases into a uniform information model and retrieval system. The actual databases from which records are retrieved and on which the Entrez indexes are based have different designs, based on the type of data, and reside on different machines. These will be referred to as the “source databases”. A common theme in the implementation of Entrez is that some functions are unique to each source database, whereas others are common to all Entrez databases.
A Division of Labor: Basic Principles
Some of the common routines and formats for every Entrez node include the term lists and posting files (i.e., the retrieval engine) used for Boolean queries, the links within and between nodes, and the summary format used for listing search results in which each record is called a DocSum. Generally, an Entrez query is a Boolean expression that is evaluated by the common Entrez engine and yields a list of unique ID numbers (UIDs), which identify records in an Entrez node. Given one or more UIDs, Entrez can retrieve the DocSum(s) very quickly. The links made within or between Entrez nodes from one or more UIDs is also a function across all Entrez source databases.
The software that tracks the addition of new or updated records or identifies those that should be deleted from Entrez may be unique for each source database. Each database must also have accompanying software to gather index terms, DocSums, and links from the source data and present them to the common Entrez indexer. This can be achieved through either a set of C++ libraries or by generating an XML document in a specific DTD that contains the terms, DocSums, and links. Although the common engine retrieves a DocSum(s) given a UID(s), the retrieval of a full, formatted record is directed to the source database, where software unique to that database is used to format the record correctly. All of this software is written by the NCBI group that runs the database.
This combination of database-specific software and a common set of Entrez routines and applications allows code sharing and common large-retrieval server administration but enables flexibility and simplicity for a wide variety of data sources.
Software
Although the basic principles of Entrez have remained the same for almost a decade, the software implementation has been through at least three major redesigns and many minor ones.
Currently, Entrez is written using the NCBI C++ Toolkit. The indexing fields (which for PubMed, for example, would be Title, Author, Publication Date, Journal, Abstract, and so on) and DocSum fields (which for PubMed are Author, Title, Journal, Publication Date, Volume, and Page Number) for each node are defined in a configuration file; but for performance at runtime, the configuration files are used to automatically generate base classes for each database. These are the basic pieces of information used by Entrez that can also be inherited and used by more database-specific, hand-coded features. The term indexes are based on the Indexed Sequential-Access Method (ISAM) and are in large, shared, memory-mapped files. The postings are large bitmaps, with one bit per document in the node. Depending on how sparsely populated the posting is, the bit array is adaptively compressed on disk using one of four possible schemes. Boolean operations are performed by using AND or OR postings of bit arrays into a result bit array. DocSums are small, fielded data structures stored on the same machines as the postings to support rapid retrieval.
The Web-based Entrez retrieval program, called query, is a fast cgi application that uses the Web application framework from the NCBI C++ Toolkit. One aspect of this framework is a set of classes that represents an HTML page. These classes allow the combination of static template pages, on the fly, with callbacks to class methods at tagged parts of the template. The Web page generated in an Entrez session contains elements from static templates and elements generated dynamically from common Entrez classes and from classes unique to one or a few Entrez nodes. Again, this design supports a common core of robust, common functionality maintained by one group, with support for customizations by diverse groups within NCBI.
Boolean query processing, DocSum retrieval, and other common functions are supported on a number of load-balanced “front-end” UNIX machines. Because Entrez can support session context (for example, in the use of query history, NCBI Cubby, Filters, etc.), a “history server” has been implemented on the front-end machines so that if a user is sent to machine “A” by the load balancer for their first query but to machine “B” for the second query, Entrez can quickly locate the user's query history and obtain it from machine “A”. Other than that, the front-end machines are completely independent of each other and can be added and removed readily from query support. Retrieval of full documents comes from a variety of “back-end” databases, depending on the node. These might be Sybase or Microsoft SQL Server relational databases of a variety of schemas or text files of various formats. Links are supported using the Sybase IQ database product.
References
- 1.
- Fishel R , Lescoe MK , Rao MR , Copeland NG , Jenkins NA , Garber J , Kane M , Kolodner R . The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. [PubMed: 8252616]
- The Entrez Search and Retrieval System - The NCBI HandbookThe Entrez Search and Retrieval System - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...