

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
McEntyre J, Ostell J, editors. The NCBI Handbook [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2002-.

This publication is provided for historical reference only and the information may be out of date.

Summary
There are many different approaches to starting a genomic analysis. These include literature searching, searching databases for gene names and other genomic features, performing sequence comparisons, or using map data to find gene information by position relative to other landmarks. The NCBI Map Viewer has been developed to facilitate this latter approach.
The purpose of this chapter is to provide a foundation for gaining maximum benefit from using the Map Viewer and related resources at NCBI. It is important to note that in this document, the term “map” refers to a position of a particular type of object in a particular coordinate system. This means, for example, that there is not one sequence map but a set of maps in sequence coordinates. Readers interested in precisely how sequence-based maps are annotated and assembled should refer to Chapter 14.
Introduction
First launched with the release of the sequence of Drosophila melanogaster in March 2000, Map Viewer is now used to present genetic, radiation hybrid (RH), cytogenetic, breakpoint, sequence-based, and clone maps for many genomes. The availability of whole genome sequences means that objects such as genes, markers, clones, sites of variation, and clone boundaries can be positioned by aligning defining sequence from these objects against the genomic sequence. This position information can then be compared to information about order obtained by other means, such as genetic or physical mapping. The results of sequence-based queries (e.g., BLAST) can also be viewed in genomic context. Our view of the genomes of a variety of organisms is constantly being improved through the increase in underlying data.
Map Viewer integrates map and sequence data from a variety of sources. The basic architecture and principle of Map Viewer can be applied to any complete or incomplete genome as long as map data exist to support it. Map Viewer is a powerful tool because it provides: (1) a mechanism to compare maps in different coordinate systems; (2) a robust query interface; (3) diverse options for configuring the display; (4) multiple functions to report and download maps and annotated information; (5) tools to manipulate nucleotide sequence such as ModelMaker (for constructing mRNAs from putative exon sequences); (6) connections to comprehensive data files for transfer by FTP; and (7) detailed descriptions of the objects displayed on the maps.
Maintenance of Data
Data Sources
Non-Sequence-based Maps. Sources of maps that are not based directly on sequence include published maps in genetic, radiation hybrid, cytogenetic, and ordinal coordinate systems (where ordinal refers to clone order). The primary sources of each map are described in the online help documentation of each genome-specific Map Viewer. We are indebted to the researchers who make their mapping results so freely available. When a new version of any map becomes available, the data are also updated in the appropriate NCBI database.
Sequence-based Maps. The sequence-based maps shown through Map Viewer can be supplied by external sources and/or supplied from features computed within NCBI. For example, when the annotated sequence for a complete genome is submitted to the sequence databases (GenBank/EMBL/DDBJ), a copy of the data may also be accessioned as Reference Sequences (RefSeqs; see Chapter 18). The gene, transcript, and other feature annotations of the submitted complete genome are processed for display in the Map Viewer. NCBI staff may then calculate and display the position of other types of features, such as marker position or points of variation, as separate maps (Table 1).
Table 1
Types of Map Viewer annotation provided by NCBI.
Some of the annotation of genomic sequence carried out by NCBI is included in the genomic reference sequences (NC, NT, and NW Accession number format); however, other annotation is represented only in the Map Viewer and in the associated reports (Table 1). This latter type of annotation is based on information in several NCBI databases (Table 2) and is particularly important for attaching biological information to sequence data. Links to these resources are provided in Map Viewer to provide further information about each annotated object. It should be noted, however, that although sequence features may be placed in a genomic context automatically, there are curation steps that affect the final displays. For example, for the human and mouse genomes, sequences defining genes and pseudogenes are reviewed by collaborators and NCBI staff and, whenever possible, used as the basis of RefSeq records (NG, NM, and NR Accession number format).
Table 2
NCBI data resources used in NCBI-generated annotation.
Feature annotation is computed primarily in two ways: (1) by alignment of the defining sequence to the genome; or (2) for sequence tagged sites (STSs), by e-PCR (1). In some genomes, gene placement is based primarily on the alignment of mRNA [Expressed Sequence Tags (ESTs) and cDNAs], but only when an encoded protein is predicted. In other cases, where transcription evidence is weaker, more weight is given to identification of protein-coding regions. Gene identification is also constrained in that a known gene cannot be placed more than once in a haplotype (except for pseuodo-autosomal regions) or on an incorrect chromosome. Thus, if any reference haplotype retains inappropriately redundant sequence that encodes a gene, only one copy will be annotated as that gene. Others will be assigned interim IDs (see Chapter 14). Some ab initio methods may also be used for gene prediction. The predicted genes, as well as the mRNAs, are supplied as separate maps (gene, RNA, or GenomeScan maps).
In some cases, the position of these features may suggest the location of other genomic regions of interest. For example, the position of STS markers can help define the position of phenotypes such as quantitative trait loci (QTL). Although the best annotation of a gene or region is always through annotation by an expert researcher, automated annotation of genomes and comparison to that provided by experts can provide significant useful information. Experts interested in analyzing or assisting with genome annotation should contact us at vog.hin.mln.ibcn@ofni.
Relationships among Coordinate Systems
In addition to supporting the display of multiple maps in the same coordinate system (e.g., multiple sequence-based maps), Map Viewer also displays maps in different coordinate systems by calculating the correspondances among them (e.g., sequence to genetic). This is accomplished by: (a) identifying features that have been placed on maps in different coordinate systems; and (b) using general conversion factors. In the first case, placement of STSs on the genome is critical for the integration of sequence data with other, non-sequence-based maps, such as genetic and RH maps. The integration of cytogenetic data with sequence data is achieved through alignment of sequence from clones that have been placed cytogentically, such as the human fluorescence in situ hybridization (FISH)-mapped clones from the Bacterial Artificial Chromosome (BAC) Resource Consortium (2). The integration of non-sequence-based maps with the sequence provides a powerful mechanism to access portions of sequence on the basis of marker or cytogenetic data. Many features, such as Single Nucleotide Polymorphisms (SNPs), ESTs, mRNAs, whole genome shotgun reads, and clones can be placed on the genome assembly by using standard DNA sequence alignment methods such as BLAST.
The identification of known genes within the genome assembly provides critical landmarks and functional context to the sequence data, which in turn makes it easier to traverse to other rich sources of gene and protein information, including publications, OMIM, RefSeq, Conserved Domain Database (CDD), and LocusLink.
The power of calculating correspondances between coordinate systems may be more apparent when considering a common application of Map Viewer, i.e., identifying candidate genes within a region defined by genetic markers. When markers are palced on both genetic and sequence maps, it is then possible to use the gene-related maps (gene, UniGene/EST, or ab initio predictions) to identify possible genes of interest. For more details on how to do this, see the Map Viewer Exercises in Chapter 24.
A Work in Progress
For many genomes, identifying and positioning chromosomes and genes within sequence blocks is an ongoing process. In those cases, the Map Viewer can be used to evaluate the evidence that supports the current representation of the sequence and visualize possible conflicts. Inconsistencies in map order or in the placement of any object can be seen in the Map Viewer; this is assisted in some cases by the use of color coding (Figures 1 and 2).
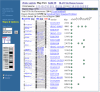
For some genomes, the color-coded contig map displays whether the annotation is based on sequence assembled from draft or finished clones (blue, finished; green, whole genome shotgun; orange, draft). This is helpful when evaluating the level of confidence in the completeness of the annotation of a gene and/or its coding region.
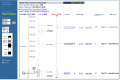
Map Viewer also uses color coding or diagrams to represent the level of confidence in the placement of any mapped object. For example, SNPs or STSs that are placed at more that one position in a given map are noted by color (yellow) in the detailed labels (Figure 3a). Annotated genes are shown in different colors, based on the source and level of confidence in the annotation or the model (Figure 3b).
Frequency of Updates
Although maps provided from external sources are updated when new data are available, the maps dependent on NCBI's annotation process are updated periodically in versions called “builds”. Thus, mRNA or other supporting evidence that becomes available after the data “freeze” date for one build will not be incorporated into the display until the next build. However, some of the supporting databases linked from the Map Viewer may have more updated information. For example, UniSTS may provide more recent e-PCR results, or LocusLink may show a newer name or additional sequence data. dbSNP may make major data releases between builds; in this case, the variation map is updated.
Methods of Access
Although most of this chapter discusses the human genome Map Viewer, there is a growing number of organisms for which there is Map Viewer access to the genome. To identify the taxa that have Map Viewer access to the genome, query the taxonomy database by typing “loprovmapviewer”[filter] into the query box on the Entrez Taxonomy homepage; or more simply, review the options provided on the Map Viewer homepage.
Links from NCBI Resources
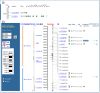
Many NCBI databases are now integrated into Map Viewer (Table 2); therefore, database records are often linked to Map Viewer displays. If a sequence in the public databases was released before the date of the current Map Viewer data freeze, then the position of this sequence may be displayed within Map Viewer. For example, Entrez Nucleotide, UniGene, UniSTS, and LocusLink records for sequences annotated on the human genome provide links directly to the appropriate region of the genome via links called Map Viewer (in the Links menu), Nucleotide, Map View, or mv links, respectively (Figure 4). It should be noted that such links are only precomputed if at least 50% of the sequence aligns with an identity of greater than 90%.
Figure 4
Connecting to Map Viewer from Entrez Nucleotide, using the Links menu in Entrez Nucleotide to connect from a record to Map Viewer.
Genome-specific resource pages also support queries via chromosome diagrams (Figure 5).

Sequence Similarity Searches
Genome-specific BLAST pages that restrict a search to a specific genome are provided for several organisms and allow the results of the search to be displayed in a genomic context (provided by Map Viewer). Genome-specific BLAST searches can be accessed from the BLAST homepage, the Map Viewer pages of individual organisms (e.g., human, mouse), and the genome-specific resource pages of individual organisms. If the reference genome (the default) is selected as the database to be searched, the Genome View button (Figure 6) will appear on the BLAST results display page.

Figure 6
Accessing the Map Viewer display from a genome-specific BLAST results page. Selecting the Genome View button shows all of the BLAST hits on the genome.
Direct Query
Simple Searches
When already at a genome-specific Map Viewer page, any combination of query terms can be entered into a Map Viewer Search for box (Figure 7). Boolean operators (AND, OR, and NOT) and the use of * as a wild card (applied to the right of any term) are supported. The Search for and Help document hyperlinks provide current details about query options. An advanced search is available for some genomes.
Queries may include any unique identifier for a database record, e.g., a sequence Accession number or OMIM (MIM) number, or a text term or phrase, e.g., a gene symbol (BRCA2) or descriptor (p53-binding), or disease name (lung cancer). The Boolean AND operator is used automatically if multiple terms are entered. Therefore, a query for “fanconi anemia” will automatically be interpreted as “fanconi AND anemia”. The wildcard operator (*) provides a convenient mechanism to retrieve genes that share a common symbol or name, as is often found for gene families. For example, a query for ABC* will return matches to the ATP-binding cassette superfamily.
The advanced query page, accessed by checking the Advanced search box, provides additional options to refine a query. These additional options, which may vary from genome to genome, are useful for restricting queries to a particular search field or map type. The advanced query page also includes predefined search options to restrict the search to data with certain properties, e.g., to only find genes associated with a known disease or with sequence variation (SNPs). Additional refinements to queries against the variation map can also be made, for example, to search for variation markers known to be in a gene or coding region.
The same options for wild cards and Boolean operators for your query term(s) apply when starting at the Map Viewer homepage. At present, however, you must select a genome to which to restrict your search. An option to query across multiple genomes is under development.
Position-based Access
To use Map Viewer to display a particular section of a genome by using a range of positions as a query, it is first necessary to select a particular chromosome for display from either a genome-specific Map Viewer page or a Genome Guide page.
Once a single chromosome is displayed, position-based queries can be defined by: (1) entering a value into the Region Shown box. This could be a numerical range (base pairs are the default if no units are entered), the names of clones, genes, markers, SNPs, or any combination. The screen will be refreshed with only that region shown. If the first entry cannot be resolved, the display will extend to the top of the map; if the second entry cannot be resolved, the display will extend to the bottom of the map. Both of these navigational aids are found on the left of the page; and (2) using the Maps & Options controls. One of the options in this menu is to define the region shown. Here it may be clearer that the region selected will be in the coordinates of the rightmost, or Master, map, which may also be adjusted in this menu. The values that can be used to specify the range are the same as those described in (1), above. (See Customizing the Display for more details on fine-tuning.)
Tutorials in Chpater 23, particularly #2, provide more examples of querying Map Viewer by position.
Interpreting the Display
Map Viewer Summary Results
The results from a query are displayed both graphically and in a summary table (Figure 8). When the query is executed by BLAST, the graphical view is color-coded according to the BLAST score, and the table summarizes the scores and the RefSeq Accession numbers that have matches. Clicking on the RefSeq Accession number (i.e., those beginning with NT_ or NW_) displays that BLAST result in the Map Viewer.
Viewing the Maps
The Graphical Display
Text or Position Queries
General information on the chromosome being viewed is summarized at the top of the map page: the species and chromosome currently being viewed, the query term, and the name of the focal map, termed the Master Map (Figure 9c).
The summary also includes the following statistics concerning the number of objects on the Master Map, which are:
- the number of objects localized (positioned) on the chromosome
- the number of objects not localized but present on the chromosome
- the number of objects localized in the region displayed (i.e., the number decreases as you zoom in)
- the number of objects for which text descriptions are shown (dependent on user-defined page length)
A thumbnail map on the left of the page provides a coarse indication of the region displayed; by default, this is a cytogenetic map, although the Master Map can be selected (Figure 9b).
Maps are displayed vertically, with the name of each map hyperlinked to a description of it (Figure 9d). Features displayed on the Master Map have brief descriptive labels; information on features on the non-Master Maps can be found by mousing over an object. The labels on the Master Map depend on the type of object and genome being explored but can provide: (a) links to resources defining the mapped element, some of which may not be at NCBI; (b) indicators of the confidence in the placement or naming or sequence in the region; (c) biological features of the element (for SNPs, this includes position in a gene or effects on the coding region); (d) direction of transcription for genes; and (e) links to tools to facilitate reviewing of the sequence (sv), downloading a subsequence of interest (seq), the mRNA alignments in a region (ev), homology maps (hm), or to create cDNA sequences in real time (mm). (See the section on Associated Tools for more information.).
Sequence (BLAST) Queries
The positions of BLAST hits are highlighted on the Contig map, and a text summary of the BLAST hit is provided with links to regional alignment reports. All of the options described previously for configuring your display are still available. Thus, it is possible to evaluate the sequence match by the location (possible intron/exon structure, percent identity) as well as to determine whether the matching genomic region contains all of the query sequence in the expected order. Adding other maps to the display using the Maps&Options window provides a powerful mechanism to determine how the query sequence corresponds to existing annotation, such as genes, gene predictions, STS markers, or SNPs. For more hints, see the tutorial section on querying the human genome by sequence.
The Tabular Display (View Data as Table/Download)
A tabular report of the region and maps being displayed can be generated by selecting the Data as Table View link (Figure 9b). The default report is restricted to maps that were in the previous graphical display. Tables indicating the object name, or other identifier, and chromosome coordinates are provided for each map, along with many of the links seen in the graphical display. If the region being displayed on the map includes more than 1000 features per map, a warning message is displayed that points to the FTP site as an alternative for large-scale access.
If any of the maps are in sequence coordinates, an option is presented to report data for any sequence map in the region. Note: Links are provided for downloading tab-delimited files for any or all maps.
Customizing the Display
The Map Viewer display can be customized with regard to the region shown, the number and coordinate systems of maps, the number of objects labeled on the Master Map, and whether to show connections between objects. Each of these will be described in this section.
Selecting the Region to Display
The Map Viewer provides zoom, navigation, and other map display controls. These can be found on the display page itself and in the Maps&Options window (Figure 10).
As the resolution of a view is changed, the chromosome diagram is updated. The view automatically centers on a highlighted query term, or on the middle of the chromosome if browsing only. The chromosome view can be moved up and down or zoomed in and out. Zooming can be achieved in several ways: (1) by using the zoom control, located in the left column; (2) by providing a range or bounding markers in the Region Shown text boxes; or (3) by selecting part of the map to display a menu with predefined zoom levels. Most menu-based zooms should be carried out in two or three steps to avoid missing the region of interest. It is also possible to scroll or reposition the display by selecting recenter from the menu that pops up when you click on the chromosome diagram at the left or on a map or by clicking on the arrows at the top and bottom of a map.
Selecting the Maps (Tracks) to Display
Maps are categorized by the coordinate system as well as type of feature. The maps available for a genome can be seen by scrolling through the Maps menu in the Maps&Options window or in the genome-specific help documentation. For display and query purposes, different types of features annotated on sequence coordinates are treated as different maps. The maps in sequence coordinates are comparable because all of the sequence maps are based on the reference to a standard genome assembly. Thus, one can display the SNP map (at high zoom level) next to the Gene, UniGene, or GenomeScan map to ascertain the number and location of polymorphisms in a region.
Some basic map controls are available directly on the display including removal of a map from the display by clicking on the X over the map and moving a secondary map to the Master Map position by clicking on the arrow next to the map label.
The Maps&Options window provides advanced options to: (a) add a ruler to any map; (b) reset the page length to display more (or less) information; (c) define region to display by providing coordinates or marker name in Region Shown boxes (also available directly on the Map Viewer display); (d) display direct connections between maps by checking the Show connections box; (e) optionally view text in Verbose or Condensed mode by selecting the checkbox. These user-defined preferences will be maintained for additional queries on different regions or chromosomes, until reset.
There has been considerable effort to integrate data on the sequence-based maps with data from non-sequence-based maps. Map connections provide a unique and powerful mechanism to identify features in a relevant region of the sequence map when starting with information from a different coordinate system (see Relationships among Coordinate Systems).
The features that are available with Map Viewer are summarized in Box 1.
Box 1
Map Viewer-associated functions.
Associated Tools
Map Viewer provides links to several tools to display, download, or manipulate the sequence in a user-defined region. Whenever a sequence-based map is the master (the one at the right), the link Download/View Sequence/Evidence is provided above the map display. This opens a window that provides access to the seq, ev, and mm tools described below. In addition, when the annotated object is a gene (sequence or cytogenetic maps) or the species-specific UniGene cluster, the label may include these links.
The Evidence Viewer (ev) displays graphically the GenBank and RefSeq cDNAs that align to the genome in a particular region, along with a density plot for ESTs. The positions of any mismatches or insertions/deletions are marked, the multiple pairwise sequence alignments are provided, and computed translations are shown.
The Sequence Viewer (sv) is the Entrez graphical display option for any nucleotide sequence, focused on the gene indicated. By default, a 2-kb section of sequence is shown below the representation of the features, but that limit can be increased at the bottom of the page. It is also possible to zoom and navigate in the display.
Sequence Download (seq) provides the same function as the Download/View Sequence link provided at the top of the Maps page. The scope of the sequence passed to the tool corresponds to what is being viewed on the page. When connected to a gene feature, the scope corresponds to that gene. The tool allows the user to alter the sequence scope and to select a report format (e.g., FASTA, GenBank, ASN.1). For the human and mouse genomes, a link is also provided to the Human–Mouse Homology Map (hm).
Model Maker (mm) displays the evidence for exons in a genomic region by diagramming the exons predicted from the alignment of cDNAs, from ab initio models (the default), and from alignment of ESTs (after an explicit selection). To facilitate construction of your own model transcript or transcripts, the splice junctions and the exons they connect are displayed, and the coding potential of any combination of exons can quickly be evaluated using ORFfinder. The sequence can also be edited, and the results can be saved or downloaded.
Technical Details
Data Access
The data displayed in Map Viewer are freely available. In addition to the view-specific reports, all of the data are available by FTP. README files document the content and format of each file. Genomic data are also available by chromosome; this includes genomic contigs (NT_ or NW_ Accession numbers) built from finished and unfinished sequence data. The contig data are available in various formats, including ASN.1, FASTA, GenBank, and GenPept. Also available in this directory are the RNAs (NM_, XM_ , and XR_ Accession numbers) and proteins (NP_, XP_).
Constructing URLs to Generate Specific Displays
Dynamic links to Map Viewer can be generated by constructing URLs with arguments that define the species, chromosome, range, types of maps (with or without units), display order, number of labels, query string, how to center a display around a query result, and the type of label for the display. The most current documentation is provided in the online help. The examples in Box 2, however, may illustrate the flexibility of the approach. Please note that the argument of the map in the URL is processed as an ordered list, with the order in the list controlling the left-to-right order in the display. Additional qualifiers control the display of a ruler and the range on the chromosome. If a query term is included as a part of the URL and that value cannot be identified on any of the maps in the list, that map will not be displayed.
Box 2
Examples of URL construction.
Implementation
Query terms are indexed for retrieval using the Entrez system. Thus, wild cards, Boolean operators, filters, and properties are managed as for other Entrez databases.
Each distinct object on the map is assigned a unique identifier that is specific to a particular build. Each object may have other secondary identifiers, such as IDs, in the sequence, Clone Repository, dbSNP, LocusLink, UniGene, or UniSTS databases. All descriptors are indexed as text. In addition, some are indexed by specific field values or by pre-identified properties, such as genes with associated diseases, SNPs with heterozygosity values in pre-defined ranges, or evidence type for genes. These field names or properties can be applied to restrict a query either in the Web-based query form or within a URL. The complete listings of current implementations for field qualifiers and properties are provided in the online help documentation.
Data for each map are retrieved for display from a relational database based on the IDs returned from the Entrez query. The database is used only to support display; it is refreshed with each NCBI build or update of any other map but not to track changes from build to build. Data from previous builds are archived at NCBI, but direct access is not currently supported.
Caveats for Using Evolving Data
Map Viewer displays represent the current synthesis of information available at the time of the data freeze (Table 3). It is important to understand that the underlying data may change from build to build, as our view of a genome becomes more refined. The data presented should always be critically reviewed, with a view to assessing the reliability of the assembly and annotation.
Table 3
Web sites of interest.
Means of reviewing reliability include: (a) noting the color coding of the contigs according to whether the sequence is draft or finished (this primarily applies to the human sequence); (b) noting the descriptions of the genes, STS, or SNPs to determine whether the element has been placed more than once; (c) checking that the STS order is the same on different maps; and (d) viewing features from different coordinate systems on the same map, e.g., showing STS features on the sequence (nucleotide coordinates), RH (cRay coordinates), and genetic maps (centiMorgan coordinates) to check for ambiguities. For more information, see the Pipeline FAQ /genome/guide/BuildFAQ.html.
References
- 1.
- Schuler GD . Electronic PCR: bridging the gap between genome mapping and genome sequencing. Trends Biotechnol. 1998;16(11):456–459. [PubMed: 9830153]
- 2.
- The BAC Resource Consortium. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature. 2001;409:953–958. [PMC free article: PMC7845515] [PubMed: 11237021]
- Using the Map Viewer to Explore Genomes - The NCBI HandbookUsing the Map Viewer to Explore Genomes - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...