

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
McEntyre J, Ostell J, editors. The NCBI Handbook [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2002-.

This publication is provided for historical reference only and the information may be out of date.

Summary
The NCBI Taxonomy database is a curated set of names and classifications for all of the organisms that are represented in GenBank. When new sequences are submitted to GenBank, the submission is checked for new organism names, which are then classified and added to the Taxonomy database. As of April 2003, there were 176,890 total taxa represented.
There are two main tools for viewing the information in the Taxonomy database: the Taxonomy Browser, and Taxonomy Entrez. Both systems allow searching of the Taxonomy database for names, and both link to the relevant sequence data. However, the Taxonomy Browser provides a hierarchical view of the classification (the best display for most casual users interested in exploring our classification), whereas Entrez Taxonomy provides a uniform indexing, search, and retrieval engine with a common mechanism for linking between the Taxonomy and other relevant Entrez databases.
Introduction
Organismal taxonomy is a powerful organizing principle in the study of biological systems. Inheritance, homology by common descent, and the conservation of sequence and structure in the determination of function are all central ideas in biology that are directly related to the evolutionary history of any group of organisms. Because of this, taxonomy plays an important cross-linking role in many of the NCBI tools and databases.
The NCBI Taxonomy database is a curated set of names and classifications for all of the organisms that are represented in GenBank. When new sequences are submitted to GenBank, the submission is checked for new organism names, which are then classified and added to the taxonomy database. As of April 1, 2003, there were 4,653 families, 26,427 genera, 130,207 species, and 176,890 total taxa represented.
Of the several different ways to build a taxonomy, our group maintains a phylogenetic taxonomy. In a phylogenetic classification scheme, the structure of the taxonomic tree approximates the evolutionary relationships among the organisms included in the classification (the “tree of life”; see Figure 1).
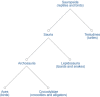
Our classification represents an assimilation of information from many different sources (see Box 1). Much of the success of the project is attributable to the flood of new molecular data that has revolutionized our understanding of the phylogeny of many groups, especially of previous poorly understood groups such as Bacteria, Archea, and Fungi. Users should be aware that some parts of the classification are better developed than others and that the primary systematic and phylogenetic literature is the most reliable information source.
Box 1
History of the Taxonomy Project.
We do not rely on sequence data alone to build our classification, and we do not perform phylogenetic analysis ourselves as part of the taxonomy project. Most of the organisms in GenBank are represented by only a snippet of sequence; therefore, sequence information alone is not enough to build a robust phylogeny. The vast majority of species are not there at all, although about 50% of the birds and the mammals are represented. We therefore also rely on analyses from morphological studies; the challenge of modern systematics is to unify molecular and morphological data to elucidate the evolutionary history of life on earth.
Adding to the Taxonomy Database
Currently, more than 100 new species are added to the database daily, and the rate is accelerating as sequence analysis becomes an ever more common component of systematic research and the taxonomic description of new species.
Sources of New Names
The EMBL and DDBJ databases, as well as GenBank, now use the NCBI Taxonomy as the standard classification for nucleotide sequences (see Box 1). Nearly all of the new species found in the Taxonomy database are via sequences submitted to one of these databases from species that are not yet represented. In these cases, the NCBI taxonomy group is consulted, and any problems with the nomenclature and classifications are resolved before the sequence entries are released to the public. We also receive consults for submissions that are not identified to the species level (e.g., “Hantavirus” or “Bacillus sp.”) and for anything that looks confusing, incorrect, or incomplete to the database indexers. All consults include information on the problem organism names, source features, and publication titles (if any). The email addresses for the submitters are also included in case we need to contact them about the nomenclature, classification, or annotation of their entries.
The number and complexity of organisms in a submission can vary enormously. Many contain a single new name, others may include 100 species, all from the same familiar genus, whereas others may include 100 names (only half identified at the species level) from 100 genera (all of which are new to the Taxonomy database) without any other identifying information at all.
Some new organism names are found by software when the protein sequence databases (SWISS-PROT, PIR, and the PRF) are added to Entrez; because most of the entries in the protein databases have been derived from entries in the nucleotide database, this is a small number. The NCBI structure group may also find new names in the PDB protein structure database. Finally, because we made the Taxonomy database publicly accessible on the Web, we have had a steady stream of comments and corrections to our spellings and classification from outside users.
More on Submission
We often receive consults on submissions with explicitly new species names that will be published as part of the description of a new species. These sequence entries (like any other) may be designated “hold until published” (HUP) and will not be released until the corresponding journal article has been published. These species names will not appear on any of our taxonomy Web sites until the corresponding sequence entries have been released.
Occasionally, the same new genus name is proposed simultaneously for different taxa; in one case, two papers with conflicting new names had been submitted to the same journal, and both had gone through one round of review and revision without detection of the duplication. Although these duplications would have been discovered in time, the increasingly common practice of including some sequence analysis in the description of a new species can lead to earlier detection of these problems. In many cases, the new species name proposed in the submitted manuscript is changed during the editorial review process, and a different name appears in the publication. Submitters are encouraged to inform us when their new descriptions have been published, particularly if the proposed names have been changed.
We strongly encourage the submission of strain names for cultured bacteria, algae, and fungi and for sequences from laboratory animals in biochemical and genetic studies; of cultivar names for sequences from cultivated plants; and of specimen vouchers (something that definitively ties the sequence to its source) for sequences from phylogenetic studies. There are many other kinds of useful information that may be contained within the sequence submission, but these data are the bare minimum necessary to maintain a reliable link between an entry in the sequence database and the biological source material.
Using the Taxonomy Browser
The Taxonomy Browser (TaxBrowser) provides a hierarchical view of the classification from any particular place in the taxonomy. This is probably the display of choice for most casual users (browsers) of the taxonomy who are interested in exploring our classification. The TaxBrowser displays only the subset of taxa from the taxonomy database that is linked to public sequence entries. About 15% of the full Taxonomy database is not displayed on the public Web pages because the names are from sequence entries that have not yet been released.
TaxBrowser is updated continuously. New species will appear on a daily basis as the new names appear in sequence entries indexed during the daily release cycle of the Entrez databases. New taxa in the classification appear in TaxBrowser on an ongoing basis, as sections of the taxonomy already linked to public sequence entries are revised.
The Hierarchical Display
The browser produces two different kinds of Web pages: (1) hierarchy pages, which present a familiar indented flatfile view of the taxonomic classification, centered on a particular taxon in the database; and (2) taxon-specific pages, which summarize all of the information that we associate with any particular taxonomic entry in the database. For example, “hominidae” as a search term from the TaxBrowser homepage finds our human family (Figure 2).
The Taxon-specific Display
The taxon-specific browser display page shows all of the information that is associated with a particular taxon in the Taxonomy database and some information collected through links with related databases (Figure 3).
There are two sets of links to Entrez records from the Taxonomy Browser. The "subtree links" are accumulated up the tree in a hierarchical fashion; for example, there are 16 million nucleotide records and a half million protein records associated with the Chordata (Figure 3a). These are all linked into the taxonomy at or below the species leves and can be retrieved en masse via the subtree hotlink.
"Direct links" will retrieve Entrez records that are linked directly to this particular node in the taxonomy database. Many of the Entrez domains (e.g., sequences and structures) are linked into the taxonomy at or below the species level; it is a data error when a sequence entry is directly linked into the taxonomy at a taxon somewhere above the species level. For other Entrez domains (e.g., literature and phylogenetic sets), this is not the case. A journal article may talk about several different species but may also refer directly to the phylum Chordata. We have searched the full text of the articles in the PubMed Central archive with the scientific names from the taxonomy database. Twenty-seven articles in PubMed Central refer directly to the phylum Chordata; 9,299 articles are linked into the taxonomy somewhere in the Chordata subtree. The PopSet domain contains population studies, phylogenetic sets, and alignments. We have recently changed the way that we index phylogenetic sets in Entrez. The five "Direct links" at the Chordata will retrieve the phylogenetic sets that explicitly span the Chordata; the "Subtree links" will also include phylogenetic sets that are completely contained within the Chordata.
The taxon-specific browser pages now also show the NCBI LinkOut links to external resources. These include links to a broad range of different kinds of resources and are provided for the convenience of our users; the NCBI does not vouch for the content of these resources, although we do make an effort to ensure that they are of good scientific quality. A complete list of external resources can be found here. Groups interested in participating in the LinkOut program should visit the LinkOut homepage.
Search Options
There are several different ways to search for names in the Taxonomy database. If the search results in a terminal node in our taxonomy, the taxon-specific browser page is displayed; if the search returns with an internal (non-terminal) node, the hierarchical classification page is displayed.
Complete Name. By default, TaxBrowser looks for the complete name when a term is typed into the search box. It looks for a case-insensitive, full-length string match to all of the nametypes stored in the Taxonomy database. For example, Homo sapiens, Escherichia, Tetrapoda, and Embryophyta would all retrieve results.
Names can be duplicated in the Taxonomy database, but the taxonomy browser can only be focused on a single taxon at any one time. If a complete name search retrieves more than one entry from the taxonomy, an intermediate name selection screen appears (Figure 4). Each duplicated name includes a manually curated suffix that differentiates between the duplicated names.

Wild Card. This is a regular expression search, *, with wild cards. It is useful when the correct spelling of a scientific name is uncertain or to find ambiguous combinations for abbreviated species names. For example, C* elegans results in a list of 16 species and subspecies (Box 2). Note: there is still only one H. sapiens.
Box 2
TaxBrowser results from using the wild card search, C* elegans.
Token Set. This treats the search string as an unordered set of tokens, each of which must be found in one of the names associated with a particular node. For example, "sapiens" retrieves:
Homo sapiens Homo sapiens neanderthalensis
Phonetic Name. This search qualifier can be used when the user has exhausted all other search options to find the organism of interest. The results using this function can be patchy, however. For example, “drozofila” and “kaynohrhabdietees” retrieve respectable results; however, “seenohrabdietees” and “eshereesheeya” are not found.
Taxonomy ID. This allows searching by the numerical unique identifier (taxid) of the NCBI Taxonomy database, e.g., 9606 or 666.
How to Link to the TaxBrowser
There is a help page that describes how to make hyperlinks to the Taxonomy Browser pages.
The Taxonomy Database: TAXON
The NCBI Taxonomy database is stored as a SyBase relational database, called TAXON. The NCBI taxonomy group maintains the database with a customized software tool, the Taxonomy Editor. Each entry in the database is a “taxon”, also referred to as a “node” in the database. The “root node” (taxid1) is at the top of the hierarchy. The path from the root node to any other particular taxon in the database is called its “lineage”; the collection of all of the nodes beneath any particular taxon is called its “subtree”. Each node in the database may be associated with several names, of several different nametypes. For indexing and retrieval purposes, the nametypes are essentially equivalent.
The Taxonomy database is populated with species names that have appeared in a sequence record from one of the nucleotide or protein databases. If a name has ever appeared in a sequence record at any time (even if it is not found in the current version of the record), we try to keep it in the Taxonomy database for tracking purposes (as a synonym, a misspelling, or other nametype), unless there are good reasons for removing it completely (for example, if it might cause a future submission to map to the wrong place in the taxonomy).
Taxids
Each taxon in the database has a unique identifier, its taxid. Taxids are assigned sequentially. When a taxon is deleted, its taxid disappears and is not reassigned (Table 1; see the FTP for a list of deleted taxids). When one taxon is merged with another taxon (e.g., if the names were determined to be synonyms or one was a misspelling), the taxid of the node that has disappeared is listed as a “secondary taxid” to the taxid of the node that remains (see the merged taxid file on the FTP site). In either case, the taxid that has disappeared will never be assigned to a new entry in the database.
Table 1
Files on the taxonomy FTP site.
Nomenclature Issues
TAXON Nametypes
There are many possible types of names that can be associated with an organism taxid in TAXON. To track and display the names correctly, the various names associated with a taxid are tagged with a nametype, for example “scientific name”, “synonym”, or “common name”. Each taxid must have one (and only one) scientific name but may have zero or many other names (for example, several synonyms, several common names, along with only one “GenBank common name”).
When sequences are submitted to GenBank, usually only a scientific name is included; most other names are added by NCBI taxonomists at the time of submission or later, when further information is discovered. For a complete description of each nametype used in TAXON, see Appendix 1.
Classes of TAXON Scientific Names
Scientific names, the only required nametype for a taxid, can be further qualified into different classes. Not all “scientific names” that accompany sequence submissions are true Linnaean Latin binomial names; if the taxon is not identified to the species level, it is not possible to assign a binomial name to it. For indexing and retrieval purposes, TAXON needs to know whether the scientific name is a Latin binomial name, or otherwise. A full listing of the classes of TAXON scientific names can be viewed in Appendix 2.
Duplicated Names
The treatment of duplicated names was discussed briefly in the section on the Taxonomy browser. For our purposes, there are four main classes of duplicated scientific names: (1) real duplicate names, (2) structural duplicates, (3) polyphyletic genera, and (4) other duplicate names.
Real Duplicate Names
There are several main codes of nomenclature for living organisms: the Zoological Code (International Code of Zoological Nomenclature, ICZN; for animals), the Botanical Code (International Code of Botanical Nomenclature, ICBN; for plants), the Bacteriological Code (International Code of Nomenclature of Bacteria, ICNB; for prokaryotes), and the Viral Code (International Code of Virus Classification and Nomenclature, ICVCN; for viruses). Within each code, names are required to be unique. When duplicate names are discovered within a code, one of them is changed (generally, the newer duplicate name). However, the codes are complex, and not all names are subject to these restrictions. For example, Polyphaga is both a genus of cockroaches and a suborder of beetles, and the damselfly genus Lestoidea is listed within the superfamily Lestoidea.
There is no real effort to make the scientific names of taxa unique among Codes, and among the relatively small set of names represented in the NCBI taxonomy database (20,000 genera), there are approximately 200 duplicate names (or about 1%), mostly at the genus level.
Early in 2002, the first duplicate species name was recorded in the Taxonomy database. Agathis montana is both a wasp and a conifer. In this case, we have used the full species names (with authorities) to provide unambiguous scientific names for the sequence entries. (The conifer is listed as Agathis montana de Laub; the wasp, Agathis montana Shest).
Structural Duplicates
In the Zoological and Bacteriological Codes, the subgenus that includes the type species is required to have the same name as the genus. This is a systematic source of duplicate names. For these duplicates, we use the associated rank in the unique name, e.g., Drosophila <genus> and Drosophila <subgenus>. Duplicated genera/subgenera also occur in the Botanical Codes, e.g. Pinus <genus> and Pinus <subgenus>.
Polyphyletic Genera
Certain genera, especially among the asexual forms of Ascomycota and Basidiomycota, are polyphyletic, i.e., they do not share a common ancestor. Pending taxonomic revisions that will transfer species assigned to “form” genera such as Cryptococcus to more natural genera, we have chosen to duplicate such polyphyletic genera in different branches of the Taxonomy database. This will maintain a phylogenetic classification and ensure that all species assigned to a polyphyletic genus can be retrieved when searching on the genus. Therefore, for example, the basidiomycete genus Sporobolomyces is represented in three different branches of the Basidiomycota: Sporobolomyces <Sporidiobolaceae>, Sporobolomyces <Agaricostilbomycetidae>, and Sporobolomyces <Erythrobasidium clade>.
Other Duplicate Names
We list many duplicate names in other nametypes (apart from our preferred “scientific name” for each taxon). Most of these are included for retrieval purposes, common names or the names of familiar paraphyletic taxa that we have not included in our classification, e.g. Osteichthyes, Coelenterata, and reptiles.
Other TAXON Data Types
Aside from names, there are several optional types of information that may be associated with a taxid. These are (1) rank, such as species, genus or family; (2) genetic code, for translating proteins; (3) GenBank division; (4) literature citations; and (5) abbreviated lineage, for display in GenBank flat files. For more details on these data types, see Appendix 3.
Taxonomy in Entrez: A Quick Tour
The TAXON database is a node within the Entrez integrated retrieval system (Chapter 15) that provides an important organizing principle for other Entrez databases. Taxonomy provides an alternative view of TAXON to that of TaxBrowser. Entrez adds some very powerful capabilities (for example, Boolean queries, search history, and both internal and external links) to TAXON, but in many ways it is an unnatural way to represent such hierarchical data in Taxonomy. (TaxBrowser is the way to view the taxonomy hierarchically.)
Taxonomy was the first Entrez database to have an internal hierarchical structure. Because Entrez deals with unordered sets of objects in a given domain, an alternative way to represent these hierarchical relationships in Entrez was required (see the section Hierarchy Fields, below).
The main focus of the Entrez Taxonomy homepage is the search bar but also worth noting are the Help and TaxBrowser hotlinks that lead to Entrez generic help documentation and the Taxonomy browser, respectively.
The default Entrez search is case insensitive and can be for any of the names that can be found in the Taxonomy database. Thus, any of the following search terms, Homo sapiens, homo sapiens, human, or Man, will retrieve the node for Homo sapiens.
As for other Entrez databases, Taxonomy supports Boolean searching, a History function, and searches limited by field. The Taxonomy fields can be browsed under Preview/Index, some are specific to Taxonomy (such as Lineage or Rank), and others are found in all Entrez databases (such as Entrez Date).
Each search result, listed in document summary (DocSum) format, may have several links associated with it. For example, for the search result Homo sapiens, the Nucleotide link will retrieve all the human sequences from the nucleotide databases, and the Genome link will retrieve the human genome from the Genomes database.
Search Tips and Tricks
A helpful list follows:
1. A search for Hominidae retrieves a single, hyperlinked entry. Selecting the link shows the structure of the taxon. On the other hand, a search for Hominidae[subtree] will retrieve a nonhierarchical list of all of the taxa listed within the Hominidae.
2. A search for species[rank] yields a list of all species in the Taxonomy database (108,020 in May 2002).
3. Find the Taxonomy update frequency by selecting Entrez Date from the pull-down menu under Preview/Index, typing “2002/02” in the box and selecting Index. The result:
2002/01 (5176) 2002/01/03 (478) 2002/01/08 (2) 2002/01/10 (2260) 2002/01/14 (7) 2002/01/16 (239)
shows that in January 2002, 5,176 new taxa were added, the bulk of which appeared in Entrez for the first time on January 10, 2002. These taxa can be retrieved by selecting 2002/01/10, then selecting the AND button above the window, followed by Go.
4. An overview of the distribution of taxa in the DocSum list can be seen if Summary is changed to Common Tree, followed by selecting Display.
5. To filter out less interesting names from a DocSum list, add some terms to the query, e.g., 2002/01/10[date] NOT uncultured[prop] NOT unspecified[prop].
Displays in Taxonomy Entrez
There are a variety of choices regarding how search results can be displayed in Taxonomy Entrez.
Summary. This is the default display view. There are as many as four pieces of information in this display, if they are all present in the Taxonomy database: (1) scientific name of the taxon; (2) common name, if one is available; (3) taxonomic rank, if one is assigned; and (4) BLAST name, inherited from the taxonomy, e.g., Homo sapiens (human), species, mammals.
Brief. Shows only the scientific names of the taxa. This view can be used to download lists of species names from Entrez.
Tax ID List. Shows only the taxids of the taxa. This view can be used to download taxid lists from Entrez.
Info. Shows a summary of most of the information associated with each taxon in the Taxonomy database (similar to the TaxBrowser taxon-specific display; Figure 3). This can be downloaded as a text file; an XML representation of these data is under development.
Common Tree. A special display that shows a skeleton view of the relationships among the selected set of taxa and is described in the section below.
LinkOut. Displays a list of the linkout links (if any) for each of the selected sets of taxa (see Chapter 17).
Entrez Links. The remaining views follow Entrez links from the selected set of taxa to the other Entrez databases (Nucleotide, Protein, Genome, etc.) The Display view allows all links for a whole set of taxa to be viewed at once.
The Common Tree Viewer
The Common Tree view shows an abbreviated view of the taxonomic hierarchy and is designed to highlight the relationships between a selected set of organisms. Figure 5 shows the Common Tree view for a familiar set of model organisms.
If there are more than few dozen taxa selected for the common tree view, the display becomes visually complex and generally less useful. When a large list of taxa is sent to the Common Tree display, a summary screen is displayed first. For example, we currently list 727 families in the Viridiplantae (plants and green algae) (Figure 6).
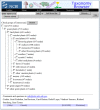
There are several formatting options for saving the common tree display to a text file: text tree, phylip tree, and taxid list.
Hyperlinks to a common tree display can be made in two ways: (1) by specifying the common tree view in an Entrez query URL (for example, this link, which displays the common tree view of all of the taxonomy nodes with LinkOut links to the Butterfly Net International Web site); or (2) by providing a list of taxids directly to the common tree cgi function (for example, this link, which will display a live version of Figure 5).
Using Batch Taxonomy Entrez
The Batch Entrez page allows you to upload a file of taxids or taxon names into Taxonomy Entrez.
Indexing Taxonomy in Entrez
As for any Entrez database, the contents are indexed by creating term lists for each field of each database record (or taxid). For TAXON, the types of fields include name fields, hierarchy fields, inherited fields, and generic Entrez fields.
Name Fields
There are five different index fields for names in Taxonomy Entrez.
All names, [name] in an Entrez search – this is the default search field in Taxonomy Entrez. This is different from most Entrez databases, where the default search field is the composite [All Fields].
Scientific name, [sname] – using [sname] as a qualifier in a search restricts it to the nametype “scientific name”, the single preferred name for each taxon.
Common name, [cname] – restricts the search to common names.
Synonym, [synonym] – restricts the search to the “synonym” nametype.
Taxid, [uid] – restricts the search to taxonomy IDs, the unique numerical identifiers for taxa in the database. Taxids are not indexed in the other Entrez name fields.
Hierarchy Fields
The [lineage] and [subtree] index fields are a way to superimpose the hierarchical relationships represented in the taxonomy on top of the Entrez data model. For an example of how to use these field limits for searching Taxonomy Entrez, see Box 3.
Box 3
Examples of combining the subtree and lineage field limits with Boolean operators for searching Taxonomy Entrez.
Lineage. For each node, the [lineage] index field retrieves all of the taxa listed at or above that node in the taxonomy. For example, the query Mammalia[lineage] retrieves 18 taxa from Entrez.
Subtree. For each node, the [subtree] index field retrieves all of the taxa listed at or below that node in the taxonomy. For example, the query Mammalia[lineage] retrieves 4,021 taxa from Entrez (as of March 9, 2002).
Next level. Returns all of the direct children of a given taxon.
Rank. Returns all of the taxa of a given Linnaean rank. The query Aves[subtree] AND species[rank] retrieves all of the species of birds with public sequence entries (there are 2,459, approximately half of the currently described species of extant birds).
Inherited Fields
The genetic code [gc], mitochondrial genetic code [mgc], and GenBank division [division] fields are all inherited within the taxonomy. The information in these fields refers to the genetic code used by a taxon or in which GenBank division it resides. Because whole families or branches may use the same code or reside in the same GenBank division, this property is usually indexed with a taxon high in the taxonomic tree, and the information is inherited by all those taxa below it. If there is no [gc] field associated with a taxon in the database, it is assumed that the standard genetic code is used. A genetic code may be referred to by either name or translation table number. For example, the two equivalent queries, standard[gc] and translation table 1[gc], each retrieves the set of organisms that use the standard genetic code for translating genomic sequences. Likewise, these two queries echinoderm mitochondrial[mgc] and translation table 9[mgc] will each retrieve the set of organisms that use the echinoderm mitochondrial genetic code for translating their mitochondrial sequences.
Generic Entrez Fields
The remaining index fields are common to most or all Entrez domains, although some have special features in the taxonomy domain. For example, the field text word, [word], indexes words from the Taxonomy Entrez name indexes. Most punctuation is ignored, and the index is searched one word at a time; therefore, the search “homo sapiens[word]” will retrieve nothing.
Several useful terms are indexed in the properties field, [prop], including functional nametypes and classifications, the rank level of a taxon, and inherited values. See Box 4 for a detailed discussion of searches using the [prop] field.
Box 4
The properties [prop] field of Taxonomy in Entrez.
More information on using the generic Entrez fields can be found in the Entrez Help documents.
Taxonomy Fields in Other Entrez Databases
Many of the Entrez databases (Nucleotide, Protein, Genome, etc.) include an Organism field, [orgn], that indexes entries in that database by taxonomic group. All of the names associated with a taxon (scientific name, synonyms, common names, and so on) are indexed in the Organism field and will retrieve the same set of entries. The Organism field will retrieve all of the entries below the term and any of their children.
To not retrieve such “exploded” terms, the unexploded indexes should be used. This query will only retrieve the entries that are linked directly to Homo sapiens: Homo sapiens[orgn:noexp]. This query will not retrieve entries that are linked to the subordinate node Homo sapiens neanderthalensis.
Taxids are indexed with the prefix txid: txid9606 [orgn].
Source organism modifiers are indexed in the [properties] field, and such queries would be in the form: src strain[prop], src variety[prop], or src specimen voucher[prop]. These queries will retrieve all entries with a strain qualifier, a variety qualifier, or a specimen_voucher qualifier, respectively.
All of the organism source feature modifiers (/clone, /serovar, /variety, etc.) are indexed in the text word field, [text word]. For example, one could query GenBank for: “strain k-12” [text word]. Because strain information is inconsistent in the sequence databases (as in the literature), a better query would be: “strain k 12”[word] OR “strain k12”[word]. Note: explicit double-quotes may be necessary for some of these queries.
The Taxonomy Statistics Page
The Taxonomy Statistics page displays tables of counts of the number of taxa in the public subset of the Taxonomy database. The numbers displayed are hyperlinks that will retrieve the corresponding set of entries. The table can be configured to display data based on three criteria: Entrez release date, rank, and taxa. The default setting shows the counts by rank for a pre-selected set of taxa (across all dates).
The checkboxes unclassified, uncultured, and unspecified will exclude the corresponding sets of taxa from the count. These work by appending the terms “NOT unclassified[prop]”, etc., to the statistics query. Checking uncultured and unspecified removes about 20% of taxa in the database and gives a much better count of the number of formally described species. As of April 1, 2003, the count was as follows:
Archaea: 82 genera, 364 species Bacteria: 1163 genera, 9927 species Eukaryota: 24939 genera, 74832 species
Selecting one of the rank categories (e.g., species) loads a new table that shows, in this example, the number of new species added to each taxon each year, starting with 1993. The Interval pull-down menu shows release statistics in finer detail. The list of taxa in the display can be customized through the Customize link.
Other Relevant References
Taxonomy FTP
A complete copy of the public NCBI taxonomy database is deposited several times a day on our FTP site. See Table 1 for details.
Tax BLAST
Taxonomy BLAST reports (Tax BLAST) are available from the BLAST results page and from the BLink pages. Tax BLAST post-processes the BLAST output results according to the source organisms of the sequences in the BLAST results page. A help page is available that describes the three different views presented on the Tax BLAST page (Lineage Report, Organism Report, and Taxonomy Report).
Toolkit Function Libraries
The function library for the taxonomy application software in the NCBI Toolkit is ncbitxc2.a (or libncbitxc2.a). The source code can be found in the NCBI Toolkit Source Browser and can be downloaded from the toolbox directory on the FTP site.
NCBI Taxonomists
In the early years of the project, Scott Federhen did all of the software and database development. In recent years, Vladimir Soussov and his group have been responsible for software and database development.
Scott Federhen (1990–present)
Andrzej Elzanowski (1994–1997)
Detlef Leipe (1994–present)
Mark Hershkovitz (1996–1997)
Carol Hotton (1997–present)
Mimi Harrington (1999–2000)
Ian Harrison (1999–2002)
Sean Turner (2000–present)
Rick Sternberg (2001–present)
Contact Us
If you have a comment or correction to our Taxonomy database, perhaps a misspelling or classification or if something looks wrong, please send a message to vog.hin.mln.ibcn@ofni.
Appendix 1. TAXON nametypes
Scientific Name
Every node in the database is required to have exactly one “scientific name”. Wherever possible, this is a validly published name with respect to the relevant code of nomenclature. Formal names that are subject to a code of nomenclature and are associated with a validly published description of the taxon will be Latinized uninomials above the species level, binomials (e.g. Homo sapiens) at the species level, and trinomials for the formally described infraspecific categories (e.g., Homo sapiens neanderthalensis). For many of our taxa, it is not possible to find an appropriate formal scientific name; these nodes are given an informal “scientific name”. The different classes of informal names are discussed in Appendix 2. Functional Classes of TAXON Scientific Names.
The scientific name is the one that will be used in all of the sequence entries that map to this node in the Taxonomy database. Entries that are submitted with any of the other names associated with this node will be replaced with this name. When we change the scientific name of a node in the Taxonomy database, the corresponding entries in the sequence databases will be updated to reflect the change. For example, we list Homo neanderthalensis as a synonym for Homo sapiens neanderthalensis. Both are in common use in the literature. We try to impose consistent usage on the entries in the sequence databases, and resolving the nomenclatural disputes that inevitably arise between submitters is one of the most difficult challenges that we face.
Synonym
The “synonym” nametype is applied to both synonyms in the formal nomenclatural sense (objective, nomenclatural, homotypic versus subjective, heterotypic) and more loosely to include orthographic variants and a host of names that have found their way into the taxonomy database over the years, because they were found in sequence entries and later merged into the same taxon in the Taxonomy database.
Acronym
The “acronym” nametype is used primarily for the viruses. The International Committee on Taxonomy of Viruses (ICTV) maintains an official list of acronyms for viral species, but the literature is often full of common variants, and it is convenient to list these as well. For example, we list HIV, LAV-1, HIV1, and HIV-1 as acronyms for the human immunodeficiency virus type 1.
Anamorph
The term “anamorph” is reserved for names applied to asexual forms of fungi, which present some special nomenclatural challenges. Many fungi are known to undergo both sexual and asexual reproduction at different points in their life cycle (so-called “perfect” fungi); for many others, however, only the asexually reproducing (anamorphic or mitosporic) form is known (in some, perhaps many, asexual species, the sexual cycle may have been lost altogether). These anamorphs, often with simple and not especially diagnostic morphology, were given Linnaean binomial names. A number of named anamorphic species have subsequently been found to be associated with sexual forms (teleomorphs) with a different name (for example, Aspergillus nidulans is the name given to the asexual stage of the teleomorphic species Emericella nidulans). In these cases, the teleomorphic name is given precedence in the GenBank Taxonomy database as the “scientific” name, and the anamorphic name is listed as an “anamorph” nametype.
Misspelling
The “misspelling” nametype is for simple misspellings. Some of these are included because the misspelling is present in the literature, but most of them are there because they were once found in a sequence entry (which has since been corrected). We keep them in the database for tracking purposes, because copies of the original sequence entry can still be retrieved. Misspellings are not listed on the TaxBrowser pages nor on the Taxonomy Entrez Info display views, but they are indexed in the Entrez search fields (so that searches and Entrez queries with the misspelling will find the appropriate node).
Misnomer
“Misnomer” is a rarely used nametype. It is used for names that might otherwise be listed as “misspellings” but which we want to appear on the browser and Entrez display pages.
Common Name
The “common name” nametype is used for vernacular names associated with a particular taxon. These may be found at any level in the hierarchy; for example, “human”, “reptiles”, and “pale devil's-claw” are all used. Common names should be in lowercase letters, except where part of the name is derived from a proper noun, for example, “American butterfish” and “Robert's arboreal rice rat”.
The use of common names is inherently variable, regional, and often inconsistent. There is generally no authoritative reference that regulates the use of common names, and there is often not perfect correspondence between common names and formally described scientific taxa; therefore, there are some caveats to their use. For scientific discourse, there is no substitute for formal scientific names. Nevertheless, common names are invaluable for many indexing, retrieval, and display purposes. The combination “Oecomys roberti (Robert's arboreal rice rat)” conveys much more information than either name by itself. Issues raised by the variable, regional, and inexact use of common names are partly addressed by the “genbank common name” nametype (below) and the ability to customize names in the GenBank flatfile.
BLAST Name
The “BLAST names” are a specially designated set of common names selected from the Taxonomy database. These were chosen to provide a pool of familiar names for large groups of organisms (such as “insects”, “mammals”, “fungi”, and others) so that any particular species (which may not have an informative common name of its own) could inherit a meaningful collective common name from the Taxonomy database. This was originally developed for BLAST, because a list of BLAST results will typically include entries from many species identified by Latin binomials, which may not be familiar to all users. BLAST names may be nested; for example, “eukaryotes”, “animals”, “chordates”, “mammals”, and “primates” are all flagged as “blast names”.
Blast names are now used in several other applications, for example the Tax BLAST displays, the Summary view in Entrez Taxonomy, and in the Summary display of the Common Tree format.
In-part
The “in-part” nametype is included for retrieval terms that have a broader range of application than the taxon or taxa at which they appear. For example, we list reptiles and Reptilia as in-part nametypes at our nodes Testudines (the turtles), Lepidosauria (the lizards and snakes), and Crocodylidae (the crocodilians).
Includes
The “includes” nametype is the opposite of the in-part nametype and is included for retrieval terms that have a narrower scope of application than the taxon at which they appear. For example, we could list “reptiles” as an “includes” nametype for the Amniota (or at any higher node in the lineage).
Equivalent Name
The “equivalent name” nametype is a catch-all category, used for names that we would like to associate with a particular node in the database (for indexing or tracking purposes) but which do not seem to fit well into any of the other existing nametypes.
GenBank Common Name
The “genbank common name” was introduced to provide a mechanism by which, when there is more than one common name associated with a particular node in the taxonomy, one of them could be designated to be the common name that should be used by default in the GenBank flatfiles and other applications that are trying to find a common name to use for display (or other) purposes. This is not intended to confer any special status or blessing on this particular common name over any of the other common names that might be associated with the same node, and we have developed mechanisms to override this choice for a common name on a case-by-case basis if another name is more appropriate or desirable for a particular sequence entry. Each node may have at most one “genbank common name”.
GenBank Acronym
There may be more than one acronym associated with a particular node in the Taxonomy database (particularly if several virus names have been synonymized in a single species). Just as with the “genbank common name”, the “genbank acronym” provides a mechanism to designate one of them to be the acronym that should be used for display (or other) purposes. Each node may have at most one “genbank acronym”.
GenBank Synonym
The “genbank synonym” nametype is intended for those special cases in which there is more than one name commonly used in the literature for a particular species, and it is informative to have both names displayed prominently in the corresponding sequence record. Each node may have at most one “genbank synonym”. For example,
SOURCE Takifugu rubripes (Fugu rubripes) ORGANISM Takifugu rubripes
GenBank Anamorph
Although the use of either the anamorph or teleomorph name is formally correct under the International Code of Botanical Nomenclature, we prefer to give precedence to the telemorphic name as the “scientific name” in the Taxonomy database, both to emphasize their commonality and to avoid having two (or more) taxids that effectively apply to the same organism. However, in many cases, the anamorphic name is much more commonly used in the literature, especially when sequences are normally derived from the asexual form of the species. In these cases, the “genbank anamorph” nametype can be used to annotate the corresponding sequence records with both names. Each node may have at most one “genbank anamorph”. For example:
SOURCE Emericella nidulans (anamorph: Aspergillus nidulans) ORGANISM Emericella nidulans
Appendix 2. Functional classes of TAXON scientific names
Formal Names
Whenever possible, formal scientific names are used for taxa. There are several codes of nomenclature that regulate the description and use of names in different branches of the tree of life. These are: the International Code of Zoological Nomenclature (ICZN), the International Code of Botanical Nomenclature (ICBN), the International Code of Nomenclature for Cultivated Plants (ICNCP), the International Code of Nomenclature of Bacteria (ICNB), and the International Code of Virus Classification and Nomenclature (ICVCN).
The viral code is less well developed than the others, but it includes an official classification for the viruses as well as a list of approved species names. Viral names are not Latin binomials (as required by the other codes), although there are some instances (e.g., Herpesvirus papio or Herpesvirus sylvilagus). When possible, we try to use ICTV-approved names for viral taxa, but new viral species names appear in the literature (and therefore in the sequence databases) much faster than they are approved into the ICTV lists. We are working to set up taxonomy LinkOut links (see Chapter 17) to the ICTV database, which will make the subset of ICTV-approved names explicit.
The zoological, botanical, and bacteriological codes mandate Latin binomials for species names. They do not describe an official classification (such as the ICTV), with the exception that the binomial species nomenclature itself makes the classification to the genus level explicit. If a genus is found to be polyphyletic, the classification cannot be corrected without formally renaming at least some of the species in the genus. (This is somewhat reminiscent of the “smart identifier” problem in computer science.)
The fungi are subject to the botanical code. The cyanobacteria (blue-green algae) have been subject to both the botanical and the bacteriological codes, and the issue is still controversial.
Authorities
“Authorities” appear at the end of the formal species name and include at least the name or standard abbreviation of the taxonomist who first described that name in the scientific literature. Other information may appear in the authority as well, often the year of description, and can become quite complicated if the taxon has been transferred or amended by other taxonomists over the years. We do not use authorities in our taxon names, although many are included in the database listed as synonyms. We have made an exception to this rule in the case of our first duplicated species name in the database, Agathis montana, to provide unambiguous names for the corresponding sequence entries.
Subspecies
All three of the codes of nomenclature for cellular organisms provide for names at the subspecies level. The botanical and bacteriological codes include the string “subsp.” in the formal name; the zoological code does not, e.g., Homo sapiens neanderthalensis, Zea mays subsp. mays, and Klebsiella pneumoniae subsp. ozaenae.
Varietas and Forma
The botanical code (but none of the others) provides for two additional formal ranks beneath the subspecies level, varietas and forma. These names will include the strings “var.” and “f.”, respectively, e.g., Marchantia paleacea var. diptera, Penicillium aurantiogriseum var. neoechinulatum, Salix babylonica f. rokkaku, or Fragaria vesca subsp. Vesca f. alba
Other Subspecific Names
We list taxa with other subspecific names where it seems useful and appropriate and where it is necessary to find places for names in the sequence databases. For indexing purposes in the Genomes division of Entrez, it is convenient to have strain-level nodes for bacterial species with a complete genome sequence, particularly when there are two or more complete genome sequences available for different strains of the same species, e.g., Escherichia coli K12, Escherichia coli O157:H7, Escherichia coli O157:H7 EDL933, Mycobacterium tuberculosis CDC1551, and Mycobacterium tuberculosis H37Rv.
Several other classes of subspecific groups do not have formal standing in the nomenclature but represent well-characterized and biologically meaningful groups, e.g., serovar, pathovar, forma specialis, and others. In many cases, these may eventually be promoted to a species; therefore, it is convenient to represent them independently from the outset, e.g., Xanthomonas campestris pv. campestris, Xanthomonas campestris pv. vesicatoria, Pneumocystis carinii f. sp. hominis, Pneumocystis carinii f. sp. mustelae, Salmonella enterica subsp. enterica serovar Dublin, and Salmonella enterica subsp. enterica serovar Panama.
Many other names below the species level have been added to the Taxonomy database to accommodate SWISS-PROT entries, where strain (and other) information is annotated with the organism name for some species.
Informal Names
In general, we try to avoid unqualified species names such as Bacillus sp., although many of them exist in the Taxonomy database because of earlier sequence entries. Bacillus sp. is a particularly egregious example, because Bacillus is a duplicated genus name and could refer to either a bacterium or an insect. In our database, Bacillus sp. is assumed to be a bacterium, but Bacillus sp. P-4-N, on the other hand, is classified with the insects.
When entries are not identified at the species level, multiple sequences can be from the same unidentified species. Sequences from multiple different unidentified species in the same genus are also possible. To keep track of this, we add unique informal names to the Taxonomy database, e.g., a meaningful identifier from the submitters could be used. This could be a strain name, a culture collection accession, a voucher specimen, an isolate name or location—anything that could tie the entry to the literature (or even to the lab notebook). If nothing else is available, we may construct a unique name using a default formula such as the submitter's initials and year of submission. This way, if a formal name is ever determined or described for any of these organisms, we can synonymize the informal name with the formal one in the Taxonomy database, and the corresponding entries in the sequence databases will be updated automatically. For example, AJ302786 was originally submitted (in November 2000) as Agathis sp. and was added to the Taxonomy database as Agathis sp. RDB-2000. In January 2002, this wasp was identified as belonging to the species Agathis montana, and the node was renamed; the informal name Agathis sp. RDB-2000 was listed as a synonym. A separate member of the genus, Agathis sp. DMA-1998, is still listed with an informal name.
Here are some examples of informal names in the Taxonomy database:
Anabaena sp. PCC 7108 Anabaena sp. M14-2 Calophyllum sp. 'Fay et al. 1997' Scutellospora sp. Rav1/RBv2/RCv3 Ehrlichia-like sp. 'Schotti variant' Gilia sp. Porter and Heil 7991 Camponotus n. sp. BGW-2001 Camponotus sp. nr. gasseri BGW-2001 Drosophila sp. 'white tip scutellum' Chrysoperla sp. 'C.c.2 slow motorboat' Saranthe aff. eichleri Chase 3915 Agabus cf. nitidus IR-2001 Simulium damnosum s.l. 'Kagera' Amoebophrya sp. ex Karlodinium micrum
We use single quotes when it seems appropriate to group a phrase into a single lexical unit. Some of these names include abbreviations with special meanings.
“n. sp.” indicates that this is a new, undescribed species and not simply an unidentified species. “sp. nr.” indicates “species near”. In the example above, this indicates that this is similar to Camponotus gasseri. “aff.”, affinis, related to but not identical to the species given. “cf.”, confer; literally, “compare with” conveys resemblance to a given species but is not necessarily related to it. “s.l.”, sensu lato; literally, “in the broad sense”. “ex”, “from” or “out of” the biological host of the specimen.
Note that names with cf., aff., nr., and n. sp. are not unique and should have unique identifiers appended to the name.
Cultured bacterial strains and other specimens that have not been identified to the genus level are given informal names as well, e.g., Desulfurococcaceae str. SRI-465; crenarchaeote OlA-6.
Names such as Camponotus sp. 1 are avoided, because different submitters might easily use the same name to refer to different species. See Box 4 for how to retrieve these names in Taxonomy Entrez.
Uncultured Names
Sequences from environmental samples are given “uncultured” names. In these studies, nucleotide sequences are cloned directly from the environment and come from varied sources, such as Antarctic sea ice, activated sewer sludge, and dental plaque. Apart from the sequence itself, there is no way to identify the source organisms or to recover them for further studies. These studies are particularly important in bacterial systematics work, which shows that the vast majority of environmental bacteria are not closely related to laboratory cultured strains (as measured by 16S rRNA sequences). Many of the deepest-branching groups in our bacterial classification are defined only by anonymous sequences from these environmental samples studies, e.g., candidate division OP5, candidate division Termite group 1, candidate subdivision kps59rc, phosphorous removal reactor sludge group, and marine archael group 1.
These samples vary widely in length and in quality, from short single-read sequences of a few hundred base pairs to high-quality, full-length 16S sequences. We now give all of these samples anonymous names, which may indicate the phylogenetic affiliation of the sequence, as far is it may be determined, e.g., uncultured archaeon, uncultured crenarchaeote, uncultured gamma proteobacerium, or uncultured enterobacterium. See Box 4 for how to retrieve these names in Taxonomy Entrez.
Candidatus Names
Some groups of bacteria have never been cultured but can be characterized and reliably recovered from the environment by other means. These include endosymbiotic bacteria and organisms similar to the phytoplasmas, which can be identified by the plant diseases that they cause. We do not give these “uncultured” names, as above. These represent a special challenge for bacterial nomenclature, because a formal species description requires the designation of a cultured type strain. The bacteriological code has a special provision for names of this sort, Candidatus, e.g., Candidatus Endobugula or Candidatus Endobugula sertula; Candidatus Phlomobacter or Candidatus Phlomobacter fragariae. These often appear in the literature without the Candidatus prefix; therefore, we list the unqualified names as synonyms for retrieval purposes.
Informal Names above the Species Level
We allow informal names for unranked nodes above the species level as well. These should all be phylogenetically meaningful groups, e.g., the Fungi/Metazoa group, eudicotyledons, Erythrobasidium clade, RTA clade, and core jakobids. In addition, there are several other classes of nodes and names above the species level that explicitly do not represent phylogenetically meaningful groups. These are outlined below.
Unclassified Bins
We are expected to add new species names to the database in a timely manner, preferably within a day or two. If we are able to find only a partial classification for a new taxon in the database, we place it as deeply as we can and list it in an explicit “unclassified” bin. As more information becomes available, these bins are emptied, and we give full classifications to the taxa listed there. In general, we suppress the names of the unclassified bins themselves so they do not appear in the abbreviated lineages that appear in the GenBank flatfiles, e.g., unclassified Salticidae, unclassified Bacteria, and unclassified Myxozoa.
Incertae Sedis Bins
If the best taxonomic opinion available is that the position of a particular taxon is uncertain, then we will list it in an “incertae sedis” bin. This is a more permanent assignment than for taxa that are listed in unclassified bins, e.g., Neoptera incertae sedis, Chlorophyceae incertae sedis, and Riodininae incertae sedis.
Mitosporic Bins
Fungi that were known only in the asexual (mitosporic, anamorphic) state were placed formerly in a separate, highly polyphyletic category of “imperfect” fungi, the Deuteromycota. Spurred especially by the development of molecular phylogenetics, current mycological practice is to classify anamorphic species as close to their sexual relatives as available information will support. Mitosporic categories can occur at any rank, e.g., mitosporic Ascomycota, mitosporic Hymenomycetes, mitosporic Hypocreales, and mitosporic Coniochaetaceae. The ultimate goal is to fully incorporate anamorphs into the natural phylogenetic classification.
Other Names
The requirement that the Taxonomy database includes names from all of the entries in the sequence database introduces a number of names that are not typically treated in a taxonomic database. These are listed in the top-level group “Other”. Plasmids are typically annotated with their host organism, using the /plasmid source organism qualifier. Broad-host-range plasmids that are not associated with any single species are listed in their own bin. Plasmid and transposon names from very old sequence entries are listed in separate bins here as well. Plasmids that have been artificially engineered are listed in the “vectors” bin.
Appendix 3. Other TAXON data types
Ranks
We do not require that Linnaean ranks be assigned to all of our taxa, but we do include a standard rank table that allows us to assign formal ranks where it seems appropriate. We do not require that sibling taxa all have the same rank, but we do not allow taxa of higher rank to be listed beneath taxa of lower rank. We allow unranked nodes to be placed at any point in our classification.
The one rank that we particularly care about is “species”. We try to ensure that all of the sequence entries map into the Taxonomy database at or below a species-level node.
Genetic Codes
The genetic codes and mitochondrial genetic codes that are appropriate for translating protein sequences in different branches of the tree of life are assigned at nodes in the Taxonomy database and inherited by species at the terminal branches of the tree. Plastid sequences are all translated with the standard genetic code, but many of the mRNAs undergo extensive RNA editing, making it difficult or impossible to translate sequences from the plastid genome directly. The genetic codes are listed on our Web site.
GenBank Divisions
GenBank taxonomic division assignments are made in the Taxonomy database and inherited by species at the terminal branches of the tree, just as with the genetic codes.
References
The Taxonomy database allows us to store comments and references at any taxon. These may include hotlinks to abstracts in PubMed, as well as links to external addresses on the Web.
Abbreviated Lineage
Some branches of our taxonomy are many levels deep, e.g., the bony fish (as we moved to a phylogenetic classification) and the drosophilids (a model taxon for evolutionary studies). In many cases, the classification lines in the GenBank flatfiles became longer than the sequences themselves. This became a storage and update issue, and the classification lines themselves became less helpful as generally familiar taxa names became buried within less recognizable taxa.
To address this problem, the Taxonomy database allows us to flag taxa that should (or should not) appear in the abbreviated classification line in the GenBank flatfiles. The full lineages are indexed in Entrez and displayed in the Taxonomy Browser.
- Summary
- Introduction
- Adding to the Taxonomy Database
- Using the Taxonomy Browser
- The Taxonomy Database: TAXON
- Nomenclature Issues
- Taxonomy in Entrez: A Quick Tour
- The Common Tree Viewer
- Indexing Taxonomy in Entrez
- The Taxonomy Statistics Page
- Other Relevant References
- NCBI Taxonomists
- Contact Us
- Appendix 1. TAXON nametypes
- Appendix 2. Functional classes of TAXON scientific names
- Appendix 3. Other TAXON data types
- The Taxonomy Project - The NCBI HandbookThe Taxonomy Project - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...