

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Molecular Imaging and Contrast Agent Database (MICAD) [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2004-2013.

| Chemical name: | [18F]-Labeled (R)-(2-(2-(2-methylpyrrolidin-1-yl)ethyl)benzofuran-5-yl)(4-fluorophenyl)-methanone |
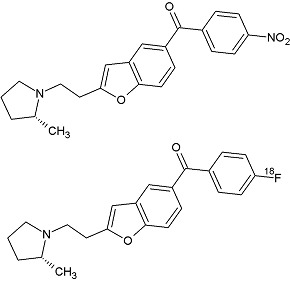
|
| Abbreviated name: | [18F]9 | |
| Synonym: | ||
| Agent Category: | Compounds | |
| Target: | Histamine subtype-3 (H3) receptor | |
| Target Category: | Receptors | |
| Method of detection: | Positron emission tomography (PET) | |
| Source of signal / contrast: | 18F | |
| Activation: | No | |
| Studies: |
| Structures of compound 12 and [18F]9 (1). |
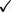 In vitro
In vitro
Background
[PubMed]
[18F]-Labeled (R)-(2-(2-(2-methylpyrrolidin-1-yl)ethyl)benzofuran-5-yl)(4-fluorophenyl)-methanone, abbreviated as [18F]9, is a 2-aminoethylbenzofuran-based histamine subtype 3 receptor (H3R) antagonist/inverse agonist that was developed for imaging H3R with positron emission tomography (PET) (1).
H3R is one of the four G-protein–coupled receptors of the histamine receptor family. The human H3R gene is located on chromosome 20q13.33, and its products are expressed predominantly in the basal ganglia, hippocampus, and cortical areas, which participate in the synthesis and release of neurotransmitters (e.g., acetylcholine, dopamine, serotonin, and noradrenaline) from histaminergic neurons (2-4). Imbalance of the histaminergic interactions results in a number of pathological states, which might be treated with H3R ligands as neuronal system modulators (5). To date, a large class of H3R ligands has been reported, which is either imidazole or non-imidazole in structure (1). These ligands have been applied to correct sleep and wakefulness disorders and narcolepsy (6). H3R antagonists have also been considered to be useful in correcting cognitive disorders and memory processes (7). However, there are many challenges for H3R ligand development because of the complexity of the central histaminergic system, the diversity of actions, and the overlapping pharmacology of H3R- and H4R-targeting compounds (5).
H3R imaging with specific radioligands has the potential to elucidate changes in the distribution and density of H3R in living human brain and to determine the dose dependence of the extent and duration of H3R occupancy by candidates. In general, non-imidazole compounds appear more promising as radiotracers for H3R imaging. In an effort to develop an 18F-labeled H3R ligand, Bao et al. selected a new chemotype compound 9, a non-imidazole 2-aminoethylbenzofuran-based H3R antagonist/inverse agonist, for imaging feasibility evaluation in animals (1). Compound 9 has previously been shown to have a high affinity and selectivity for human H3R and to be able to cross the blood−brain barrier (8). In addition, compound 9 can be labeled with no-carrier-added fluorine-18 (t1/2 = 109.7 min) at an aryl carbon. The results obtained by Bao et al. have shown that [18F]9 is an effective radioligand for H3R imaging in mice and monkeys (1).
Synthesis
[PubMed]
Compound 9 was prepared from commercially available (4-fluorophenyl)(4-hydroxyphenyl)methanone as described previously (1, 8). Bao et al. labeled compound 9 with cyclotron-produced [18F]fluoride ions through its nitro analog precursor 12 ((R)-(2-(2-(2-methylpyrrolidin-1-yl)ethyl)benzofuran-5-yl)(4-nitrophenyl)methanone) in a Synthia device equipped with a microwave heater (1). A higher decay-corrected radiochemical yield (RCY, 34%) of [18F]9 was obtained when the precursor 12 was reacted with [18F]fluoride ions in N,N-dimethylformamide than in MeCN (2%) or in dimethyl sulfoxide (6%). The reaction temperature (<90°C) was controlled by setting the microwave power between 50 W and 60 W because decomposition of [18F]9 became significant above 100°C. [18F]9 was separated with single-pass reverse-phase high-performance liquid chromatography (HPLC), with a high radiochemical purity (>99%). No residual precursor 12 or other chemical impurities were detected with HPLC in the formulated radioligand. The specific radioactivity of [18F]9 was 48.51 ± 8.18 GBq/µmol (1,311 ± 221 mCi/μmol; n = 12) at the time of intravenous injection to animals (~110 min from the end of radionuclide production). The average RCY of the formulated [18F]9 was 9.3 ± 5.8% (n = 12).
The distribution constant (log D7.4) value of [18F]9 between n-octanol and sodium phosphate buffer (0.15 M, pH 7.4) was 2.95 ± 0.06 (n = 6). The computed log D7.4 (pH 7.4) values of compounds 9 and 12 were 2.90 and 2.45, respectively. The computed partition coefficient (log P) values for compounds 9 and 12 were 4.79 ± 0.43 and 4.34 ± 0.34, respectively.
[18F]9 was found to be 98.8 ± 0.2% intact after 2.5 h in sodium phosphate buffer (pH 7.4) at room temperature (n = 6). [18F]9 was also stable in monkey whole blood (98.4 ± 0.07% intact; n = 6) and monkey plasma (99.2 ± 0.1% intact; n = 6) for 2.5 h at room temperature.
In Vitro Studies: Testing in Cells and Tissues
[PubMed]
Binding of compounds 9 and 12 to a group of human receptors was determined with competitive binding assays (no detailed description of the assays) (1). The assays confirmed the high H3R affinity of compound 9, with Ki in the nanomolar range (Table 1). The binding affinities of compound 9 for other human recombinant receptors and binding sites were at least 200-fold lower. The nitro precursor 12 had a binding affinity for human H3R almost 5-fold higher than that of compound 9 (Table 1).
Table 1. Ki values of compounds 9 and 12 as determined with competitive binding assays.
| Receptor or binding site | Ki (nM) | |
| Compound 9 | Compound 12 | |
| H1R | 5,440 | 7,055 |
| H2R | 1,708 | 923 |
| H3R | 1.9 ± 0.23 | 0.4 ± 0.04 |
| H4R | >10,000 | >10,000 |
| 5-HT1A | 432 | >10,000 |
| 5-HT1B | >10,000 | >10,000 |
| 5-HT1D | 6,380 | 5,237 |
| 5-HT2A | 2,718 | 1,355 |
| 5-HT2B | 3,152 | >10,000 |
| Dopamine active transporter | >10,000 | 137 |
| Muscarinic acetylcholine receptor M1 (M1) | 3,266 | 931 |
| M2 | 462 | 681 |
| M3 | 204 | 512 |
| M4 | 351 | 415 |
| M5 | 437 | 336 |
| All others* | >10,000 | >10,000 |
*5-HT1E,2C,3,5A,6 and 7, α1A,1B,1D,2A,2B and 2C, β1–3, D1–5, σ1,2, serotonin transporter, and norepinephrine transporter.
Animal Studies
Rodents
[PubMed]
Bao et al. first determined the time–activity curve of [18F]9 in the brain of wild-type FVB mice (1). Images were acquired at baseline ([18F]9 only; 2.7 ± 1.1 MBq (73 ± 29 μCi)) and after pretreatment (30 min before injection of [18F]9) with the selective high-affinity H3R inverse agonist ciproxifan (2.0 mg/kg), nitro precursor 12 (2.0 mg/kg), or ligand 9 itself (1.0 mg/kg) (n = 3–4 mice/group). Brain uptake of the radioactivity was expressed as standardized uptake value (SUV), where SUV = (% injected dose per cm3 brain) × (g body weight).
[18F]9 crossed the blood–brain barrier quickly, with peak whole-brain uptake of 3.36 SUV at ~6.5 min. The brain radioactivity then declined slowly to ~2.3 SUV by 90 min. In mice pretreated with ciproxifan, the brain radioactivity also quickly peaked, but at a lower level of ~1.98 SUV at 4.5 min after [18F]9 injection, and then reduced gradually to 1.09 SUV at 90 min. In mice pretreated with nitro precursor 12 or ligand 9, the [18F]9 brain radioactivity peaked at 3.5 min, with higher values of 4.34 SUV and 4.07 SUV, respectively. The brain radioactivity then decreased quickly to <1.20 SUV by 90 min. The brain radioactivity uptake was different between the pretreatment experiments with ciproxifan and those with the structural congeners 12 and 9. One possible explanation is that, at the administered doses, compounds 9 and 12 displace the structurally similar [18F]9 from plasma proteins, giving a higher plasma free fraction and greater entry of [18F]9 into brain than in the baseline experiments. Another possibility is that compounds 9 and 12, but not ciproxifan, blocks [18F]9 from binding to other unknown sites in the periphery, thereby increasing availability of [18F]9 for brain entry. Overall, these results indicate that a high proportion of brain radioactivity in the baseline experiments represented specific binding of [18F]9 to H3R.
Non-Human Primates
[PubMed]
Bao et al. then determined the time–activity curve of [18F]9 in the brains of male rhesus monkeys at baseline ([18F]9 only; 144.67 ± 17.02 MBq (3.91 ± 0.46 mCi); n = 4 monkeys) and after pretreatment with ciproxifan (2.0 mg/kg; n = 3 of the four monkeys) or compound 9 (1.0 mg/kg; n = 2 of the four monkeys) 30 min before [18F]9 injection (1). PET scans were acquired for 180 min, except for one scan of 120 min. In two of the monkeys, arterial blood was sampled at every 15 s for an initial 2 min, followed by further sampling at 3, 5, 10, 30, 60, 75, 90, 120, 150, and 180 min after [18F]9 injection. The radioactivity in each region of the brain was expressed as SUV. The two-tissue compartment model was applied to calculate the total distribution volume (VT).
[18F]9 entered the brain tissue well, with peak uptake in H3R-rich regions, such as striatum (4.45 SUV at ~27.5 min) and frontal cortex (3.86 SUV at ~47.5 min). Peak radioactivity in other regions was lower, but still higher than in cerebellum (2.76 SUV). The radioactivity in all regions decreased slowly to the end of PET scans (180 min). There was no radioactivity uptake into the skull. These results showed that the distribution of [18F]9 reflected the distribution of H3R. Pretreatment with ciproxifan reduced the peak radioactivity in H3R-rich regions, such as frontal cortex and striatum, but not in cerebellum. Subsequent washout of radioactivity from all H3R-containing regions was slow. Pretreatment with compound 9 reduced peak radioactivity in all brain regions, where the radioactivity declined to a common low level at 180 min. The summed PET images from pretreatment experiments with compound 9 showed a uniform low distribution of radioactivity across brain. Again, there was no radioactivity in the skull. Ciproxifan is known to show species differences in H3R binding affinity, with affinity for human H3R (Ki = 63 nM) >100-fold lower than that for rat H3R (Ki = 0.51 nM). The binding affinity of ciproxifan for monkey H3R is unknown. There is a possibility that the affinity of ciproxifan for monkey H3R is similar to that for human H3R, which may account for incomplete blockade of the brain H3R at the administered dose.
In the baseline experiments, the [18F]9 radioactivity cleared rapidly from plasma until ~50 min after injection, when the low level of decay-corrected plasma radioactivity became almost constant. Similar plasma time–radioactivity curves were observed in monkeys pretreated with ciproxifan or compound 9. The plasma free fraction of [18F]9 was 2.08 ± 0.14% at baseline and 1.4 ± 0.1% in monkeys pretreated with compound 9.
The [18F]9 metabolites in the monkey plasma were analyzed after extraction of the radioactivity (95.3 ± 7.44%; n = 115) from plasma with acetonitrile (1). Radio-HPLC showed that the [18F]9 intact in plasma declined continuously, while three radiometabolites [18F]A–C emerged. These radiometabolites appeared to be less lipophilic than [18F]9 (tR = 5.63 min), according to their shorter retention times during HPLC. The radiometabolites were also observed in the plasma of monkeys pretreated with ciproxifan. In monkeys pretreated with compound 9, intact [18F]9 decreased more quickly to become the minor component, and the least lipophilic radiometabolite [18F]A became the major component in plasma. The ability of the radiometabolites to penetrate the blood–brain barrier was unknown. Considering their lower lipophilicities, these radiometabolites may enter brain tissues less readily than [18F]9 and may be less troublesome for quantification of [18F]9 binding to H3R. The routes of [18F]9 metabolism and the identities of the radiometabolites were also unknown. The lack of radioactivity uptake in skull indicated that none of the radiometabolites of [18F]9 was the [18F]fluoride ion, and that radiodefluorination did not occur for [18F]9.
The brain time–activity curves in baseline monkey experiments with [18F]9 fitted well to both one-tissue and two-tissue (2TC) compartmental models (1). The F test showed that the 2TC model gave the best fit to acquired data (Table 2). On average, ciproxifan reduced the normalized total volume of distribution (VT) 26%–34%, and ligand 9 reduced VT ~49%–58%, indicating that the majority of the radioactivity in H3R-rich regions of the brain represented specific binding of [18F]9 to H3R.
Table 2. Estimation of VT from 2TC model in two monkeys.
| Region | Monkey 1 VT | Monkey 2 VT | Mean decrease (%) | |||||
| Baseline | Ciproxifan | Cold 9 | Baseline | Ciproxifan | Cold 9 | Ciproxifan | Cold 9 | |
| Frontal cortex | 64.1 | 43.5 | 29.5 | 42.1 | 26.7 | 16.4 | –34 | –58 |
| Striatum | 54.7 | 42.7 | 29.1 | 45.3 | 31.6 | 19.7 | –26 | –52 |
| Hippocampus | 60.4 | 45.3 | 31.2 | 50.8 | 30.4 | 20.8 | –33 | –54 |
| Thalamus | 53.7 | 40.8 | 29.9 | 49.9 | 29.4 | 19.4 | –33 | –53 |
| Cerebellum | 42.0 | 33.5 | 24.8 | 28.5 | 19.6 | 12.0 | –26 | –49 |
References
- 1.
- Bao X., Lu S., Liow J.S., Zoghbi S.S., Jenko K.J., Clark D.T., Gladding R.L., Innis R.B., Pike V.W. Radiosynthesis and evaluation of an (18)F-labeled positron emission tomography (PET) radioligand for brain histamine subtype-3 receptors based on a nonimidazole 2-aminoethylbenzofuran chemotype. J Med Chem. 2012;55(5):2406–15. [PMC free article: PMC3303611] [PubMed: 22313227]
- 2.
- Leurs R., Bakker R.A., Timmerman H., de Esch I.J. The histamine H3 receptor: from gene cloning to H3 receptor drugs. Nat Rev Drug Discov. 2005;4(2):107–20. [PubMed: 15665857]
- 3.
- Strakhova M.I., Fox G.B., Carr T.L., Witte D.G., Vortherms T.A., Manelli A.M., Miller T.R., Yao B.B., Brioni J.D., Esbenshade T.A. Cloning and characterization of the monkey histamine H3 receptor isoforms. Eur J Pharmacol. 2008;601(1-3):8–15. [PubMed: 18977214]
- 4.
- Chepkova A., Yanovsky E., Parmentier R., Ohtsu H., Haas H.L., Lin J.S., Sergeeva O.A. Histamine receptor expression, hippocampal plasticity and ammonia in histidine decarboxylase knockout mice. Cell Mol Neurobiol. 2012;32(1):17–25. [PubMed: 21710252]
- 5.
- Tiligada E., Kyriakidis K., Chazot P.L., Passani M.B. Histamine pharmacology and new CNS drug targets. CNS Neurosci Ther. 2011;17(6):620–8. [PMC free article: PMC6493842] [PubMed: 22070192]
- 6.
- Barbier A.J. andBradbury, M.J. Histaminergic control of sleep-wake cycles: recent therapeutic advances for sleep and wake disorders. CNS Neurol Disord Drug Targets. 2007;6(1):31–43. [PubMed: 17305552]
- 7.
- Esbenshade T.A., Browman K.E., Bitner R.S., Strakhova M., Cowart M.D., Brioni J.D. The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br J Pharmacol. 2008;154(6):1166–81. [PMC free article: PMC2483387] [PubMed: 18469850]
- 8.
- Cowart M., Faghih R., Curtis M.P., Gfesser G.A., Bennani Y.L., Black L.A., Pan L., Marsh K.C., Sullivan J.P., Esbenshade T.A., Fox G.B., Hancock A.A. 4-(2-[2-(2(R)-methylpyrrolidin-1-yl)ethyl]benzofuran-5-yl)benzonitrile and related 2-aminoethylbenzofuran H3 receptor antagonists potently enhance cognition and attention. J Med Chem. 2005;48(1):38–55. [PubMed: 15634000]
- Radiosynthesis and evaluation of an (18)F-labeled positron emission tomography (PET) radioligand for brain histamine subtype-3 receptors based on a nonimidazole 2-aminoethylbenzofuran chemotype.[J Med Chem. 2012]Radiosynthesis and evaluation of an (18)F-labeled positron emission tomography (PET) radioligand for brain histamine subtype-3 receptors based on a nonimidazole 2-aminoethylbenzofuran chemotype.Bao X, Lu S, Liow JS, Zoghbi SS, Jenko KJ, Clark DT, Gladding RL, Innis RB, Pike VW. J Med Chem. 2012 Mar 8; 55(5):2406-15. Epub 2012 Feb 16.
- Review (11)C-Labeled 6-[(3-cyclobutyl-2,3,4,5-tetrahydro-1H-3-benzazepin-7-yl)oxy]-N-methyl-3-pyridinecarboxamide hydrochloride.[Molecular Imaging and Contrast...]Review (11)C-Labeled 6-[(3-cyclobutyl-2,3,4,5-tetrahydro-1H-3-benzazepin-7-yl)oxy]-N-methyl-3-pyridinecarboxamide hydrochloride.Chopra A. Molecular Imaging and Contrast Agent Database (MICAD). 2004
- Review (11) C-labeled and (18) F-labeled PET ligands for subtype-specific imaging of histamine receptors in the brain.[J Labelled Comp Radiopharm. 2013]Review (11) C-labeled and (18) F-labeled PET ligands for subtype-specific imaging of histamine receptors in the brain.Funke U, Vugts DJ, Janssen B, Spaans A, Kruijer PS, Lammertsma AA, Perk LR, Windhorst AD. J Labelled Comp Radiopharm. 2013 Mar-Apr; 56(3-4):120-9.
- Compared pharmacology of human histamine H3 and H4 receptors: structure-activity relationships of histamine derivatives.[Br J Pharmacol. 2006]Compared pharmacology of human histamine H3 and H4 receptors: structure-activity relationships of histamine derivatives.Gbahou F, Vincent L, Humbert-Claude M, Tardivel-Lacombe J, Chabret C, Arrang JM. Br J Pharmacol. 2006 Apr; 147(7):744-54.
- Review [(18)F]3-(1H-Imidazol-4-yl)propyl-4-fluorobenzyl ether.[Molecular Imaging and Contrast...]Review [(18)F]3-(1H-Imidazol-4-yl)propyl-4-fluorobenzyl ether.Leung K. Molecular Imaging and Contrast Agent Database (MICAD). 2004
- [18F]-Labeled (R)-(2-(2-(2-methylpyrrolidin-1-yl)ethyl)benzofuran-5-yl)(4-fluoro...[18F]-Labeled (R)-(2-(2-(2-methylpyrrolidin-1-yl)ethyl)benzofuran-5-yl)(4-fluorophenyl)-methanone - Molecular Imaging and Contrast Agent Database (MICAD)
- Caenorhabditis elegans Calponin-homology (CH) domain-containing protein (fln-1),...Caenorhabditis elegans Calponin-homology (CH) domain-containing protein (fln-1), partial mRNAgi|1831512089|ref|NM_001268292.3|Nucleotide
- Anas falcata isolate IBSS-WP-138 control region, partial sequence; mitochondrialAnas falcata isolate IBSS-WP-138 control region, partial sequence; mitochondrialgi|58802724|gb|AY881800.1|Nucleotide
- Anas strepera isolate IBSS-WP-137 control region, partial sequence; mitochondria...Anas strepera isolate IBSS-WP-137 control region, partial sequence; mitochondrialgi|58802720|gb|AY881796.1|Nucleotide
- Gadolinium-1,4,7,10-tetraazacyclododecane-N',N'',N''',N''''-tetraacetic acid-Gly...Gadolinium-1,4,7,10-tetraazacyclododecane-N',N'',N''',N''''-tetraacetic acid-Gly-Pro-D-Leu-D-Ala-NHOH - Molecular Imaging and Contrast Agent Database (MICAD)
Your browsing activity is empty.
Activity recording is turned off.
See more...