

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
NCBI News [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 1991-2012.

NCBI Insights blog post: Creating custom BLAST databases
Tuesday, November 26, 2013
The latest Quick Tip blog focuses on creating custom BLAST databases, which is an easy way to speed up BLAST analysis. The blog post takes you through the process, from selecting the appropriate parent database to manipulating the filters and Entrez Query.
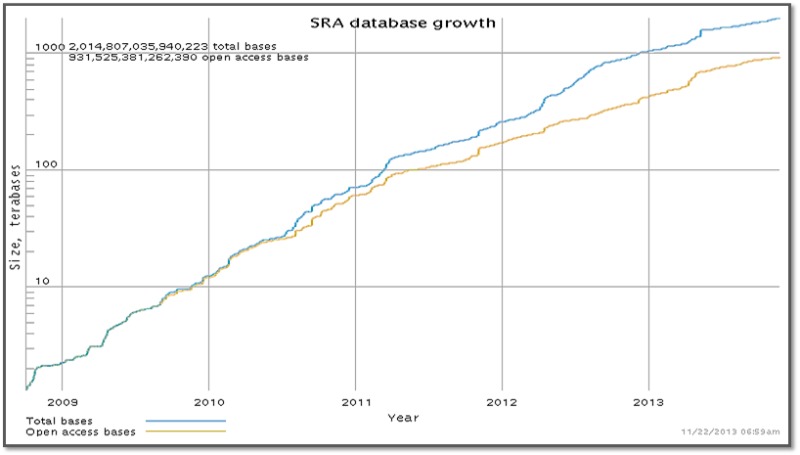
SRA milestone: Over 2 petabases of sequence data
Monday, November 25, 2013
The Short Read Archive (SRA) now contains more than 2 petabases of high-throughput sequence data. One petabase of data is open access, while the rest are sequences from 40,000 individuals who have participated in human clinical studies catalogued in dbGaP.
Planned change in bacterial strain-level information management
Thursday, November 21, 2013
Please be aware that there is an upcoming change (January 2014) in how NCBI manages organism strain information. Due to significant increases in the volume of strain-specific sequencing, we are changing our management of strain information.
Next generation sequencing has already changed the way microbial genomes are being used. The scope of microbial sequencing projects has shifted from a single isolate representing an organism to multi-isolate and multi-species projects representing microbial communities. Consequently, in the first nine months of 2013 the sequences of more than 6000 prokaryotic genomes were released by INSDC (DDBJ/ENA/GenBank).
NCBI is introducing several changes in prokaryotic genomes and related resources such as Assembly, BioProject, BioSample, and Taxonomy that will affect your submissions, data downloads, analysis tools, and parsers.
Assigning strain-level TaxID will be discontinued in January 2014 because curation of strain-level TaxIDs will not remain possible under such growth. However, the thousands of existing strain-level TaxIDs will remain, and we will continue to add informal strain-specific names for genomes from specimens that have not yet been identified to the species level, e.g. “Rhizobium sp. CCGE 510” and “Micromonas sp. RCC299”. The strain information will continue to be collected and displayed.
Submitters of genome sequences will be required to register sample meta-data in the BioSample database for each organism that they are sequencing. The BioSample submission will include the strain information and other metadata, such as culture collection and isolation information, as appropriate. The BioSample accession will be a link on the GenBank records, and the GenBank records themselves will display the strain in the source information.
Submitters of genome sequences are already required to register meta-data about the research project in the BioProject database. We no longer require a one-to-one relationship between a BioProject accession and a genome. Instead, a research effort examining multiple strains of a species or multiple species of drug-resistant bacteria, for example, could be registered as a single BioProject.
Each genome assembly is loaded to the Assembly database and assigned an Assembly accession. The Assembly accession is specific for a particular genome submission.
What defines a genome?
A BioProject ID or accession cannot be used to define a single genome, since many may belong to a multi-isolate or multi-species project. Furthermore, a TaxID can no longer reliably define an individual genome since unique TaxIDs will not be assigned for individual strains and isolates. The collection of DNA sequences of an individual sample (isolate) will be represented by a BioSample accession and if raw sequence reads are assembled and submitted to GenBank they will get a unique Assembly accession. The Assembly accession is specific for a particular genome submission. For example, sequence data generated from a single sample (with a BioSample accession) could be assembled with two different algorithms and so have two sets of GenBank accessions, each with its own Assembly accession.
For example, BioProject PRJNA203445 is a multi-species project with multiple strains and isolates of different food pathogens. Each isolate has its own BioSample accession and each assembled genome has its own Assembly accession. This BioProject includes an isolate of Listeria monocytogenes (TaxID 1639, strain R2-502) which was registered as BioSample SAMN02203126, and its genome is represented in GenBank records CP006595-CP006596, which are tracked as a group in the Assembly database under accession GCA_000438585.
FTP files
Genome text reports on the FTP site have been modified to include the BioSample and Assembly accessions. These two columns were added at the end of the tables to minimize problems for existing parsers. Initially, not all assemblies will have a BioSample accession because we are still in the process of back-filling BioSamples for genomes.
These changes will occur in January 2014. We will be releasing more information as the date approaches.
Exploring next-gen sequencing experiments with SRA-BLAST
Tuesday, November 19, 2013
NCBI’s SRA-BLAST has two new features that substantially improve its ability to explore the myriad of next-gen sequencing studies available from NCBI’s Sequence Read Archive (SRA).
First, we have expanded SRA-BLAST to include technologies beyond 454, which means that more than 100,000 experiments are currently available through this service. Except for two types of data -- human data with controlled access that are only available through dbGaP and reads stored as alignment references (cSRA format)-- experiments that can be searched through BLAST now include data from all current next-sequencing technologies that produce read lengths long enough (approximately 100 bases) for general BLAST searching.
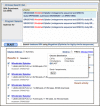
The other new feature is that SRA-BLAST now offers two different ways of finding data sets to search. The BLAST service itself provides an autocomplete feature under “Choose Search Set” that finds matches to experiment, study and run accessions as well as text from experiment descriptions (Figure 1, top panel). You can now also use the Entrez SRA system to identify experiments of interest and load these as BLAST databases in SRA BLAST through the ‘Send to’ menu from the SRA search results (Figure 1, bottom panel).
Example
There are many studies in SRA in which the experiments form a series with varying conditions. We can use SRA BLAST to examine changes in the data under these different conditions. For example, let’s look at study SRP001041, which contains metagenomic sequence data from a depth profile of the North Pacific subtropical gyre from station ALOHA with samples from 25, 75, 110 and 500 meters (SRX007372, SRX007369, SRX007370, and SRX007371). (See the Hawaii Ocean Time Series website for details on the sampling location and projects there.) We can easily find these experiments using the following search in SRA, which retrieves the four DNA-based experiments from the different depths.
SRP001041 AND dna data[Filter]
Using SRA BLAST we can profile the abundance of Prochlorococcus, a tiny prokaryotic photosynthetic organism that plays a large role in carbon cycling in the open ocean (reviewed in Partensky F, Hess WR, and Vaulot D, 1999, PCMID: 98958).
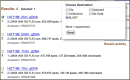
After selecting one of the four experiments, for example the one from 25 meters, we can load it as a BLAST database through the ‘Send to’ menu (Figure 2).
Figure
Figure 2. Entrez SRA summaries of four metagenomic experiments from a vertical profile of the North Pacific Ocean. One or more selected experiments may be set as BLAST databases for searching through the ‘Send to’ menu.
We can then use the coding region of one the genes involved in Prochlorococcus photosynthesis to measure the abundance of these organisms at different depths. The photosystem I P700 chlorophyll a apoprotein A1 (psaA) region (CP000551.1|:c1473975-1471672)Prochlorococcus marinus str. AS9601 serves as a useful marker query. The following link will set up a BLAST search with the gene region against the 25-meter data.
Figure 3 shows the BLAST results for the psaA query at different depths indicating the decline in abundance of the organisms with depths below 100 meters.

The SRA-BLAST service combined with the new ‘Send to’ feature in the Entrez SRA database provides a convenient and interesting way to explore the many datasets now in NCBI’s Sequence Read Archive.
NCBI Insights blog post: Saved Searches and E-mail Alerts
Monday, November 18, 2013
As part of the My NCBI service, PubMed and other Entrez databases allow users to save searches and then receive regular e-mail alerts about new records retrieved by that search. Please see the new NCBI Insights blog post for details about setting up these searches and alerts.
For more information, see the following:
RefSeq release 62 now available
Monday, November 18, 2013
The complete RefSeq release 62 is now available with nearly 50 million records describing more than 36,036,343 proteins and 5,178,509 transcripts from 31,646 different organisms. More details about RefSeq release 62 are in the release statistics and the release notes.
NCBI's Eukaryotic Annotation Pipeline has now annotated the genomes for 100 different organisms!
Tuesday, November 12, 2013
NCBI began annotating eukaryotic genomes in 2000. We have now completed the genome annotation for 100 different organisms, including 50 mammals, 25 other vertebrates, 17 invertebrates and 8 plants. Among these 100 organisms, 47 were annotated for the first time in 2013. The lucky 100th organism is the Chinese alligator (Alligator sinensis).
Recent improvements in the Eukaryotic Genome Annotation Pipeline have not only increased the throughput but have also improved the quality of the annotation produced. For example, incorporation of RNASeq data for use in gene prediction has permitted the annotation of organisms with little traditional transcript or protein sequence. View annotation runs recently completed or in progress.
Data produced by the Eukaryotic Genome Annotation Pipeline is available in the Reference Sequences (RefSeq) collection, BLAST non-redundant and organism-specific databases, Gene database, and on the NCBI FTP site.
Need a public genome annotated? Make a request!
NCBI's 25th Anniversary and The Jim Gray eScience Award
Tuesday, November 05, 2013
November 2013 marks 25 years since the founding of the National Center for Biotechnology Information (NCBI).
In honor of NCBI’s 25th anniversary, United States Senator Ben Cardin read a statement into the Congressional Record recognizing years of service in providing access to biomedical and genomic information to enhance the world’s science and health.

In addition, on November 1st, an awards and recognition program was held to commemorate this occasion. At this event, Tony Hey, Ph.D., Vice President of Microsoft Research, presented NCBI Director David Lipman, M.D., with the Jim Gray eScience Award which recognizes outstanding contributions to the field of data-intensive computing in the pursuit of open, supportive, and collaborative research models.
For more information, see this Microsoft Research Connections Blog post.

Figure
Tony Hey, Ph.D., Vice President of Microsoft Research, presents the Jim Gray eScience award to David Lipman, M.D., Director of the NCBI.
The NCBI awards program also featured presentations by:
- Michael M. Gottesman, M.D., NIH Deputy Director for Intramural Research - Introductory Remarks
- Donald A.B. Lindberg, M.D., Director of the NLM - Recollections on the origins of the NCBI
- Sir Richard J. Roberts, Ph.D, Chief Scientific Officer of New England Biolabs - Keynote Address: “A personal recollection of GenBank and NCBI”

Figure
Michael Gottesman, M.D., NIH Deputy Director for Intramural Research, Sir Richard Roberts, Ph.D., Chief Scientific Officer of New England Biolabs and David Lipman, M.D., Director of the NCBI.
New SNP data available for several organisms!
Monday, November 04, 2013
New SNP data (build 139) is now available on the web and in FTP files for several organisms, including gorilla, horse, dog, sheep, rabbit, opossum, platypus, wild turkey, zebra finch, tomato, grape and aspergillus.
Update on PubMed Commons' comments in the early pilot phase
Friday, November 01, 2013
PubMed Commons is a new system that enables researchers to share their opinions about scientific publications indexed in the PubMed database. Participation in PubMed Commons requires users with My NCBI accounts to join before they can view or add comments.
As of November 1, 2013, there are about 1,000 people signed up in the Commons and in just four days of public access the amount of comments on PubMed records doubled to over 200.
Approximately a third of the first ~200 comments included critique or pointed to other studies or reviews with the potential to change people’s interpretations or conclusions. Some authors posted corrections or changed their own conclusions in the light of others’ subsequent work. Authors also used PubMed Commons to update people on their work – including links to databases that have moved, providing contextual information and backstories as well as new, relevant work.
Many PubMed Commons participants took the opportunity to add links to relevant papers and data, sometimes in the non-PubMed academic literature or data repositories – including complete datasets, data re-analyses, blog posts and full text pre-prints of the article.
Around half of the comments were principally discussion, developing lines of thoughts and raising or asking questions and there has already been some interesting back and forth between PubMed Commons participants interested in an issue and authors of the PubMed records.
For more information, please see:
NCBI Insights Blog Posts:
- NCBI Insights blog post: Creating custom BLAST databases
- SRA milestone: Over 2 petabases of sequence data
- Planned change in bacterial strain-level information management
- Exploring next-gen sequencing experiments with SRA-BLAST
- NCBI Insights blog post: Saved Searches and E-mail Alerts
- RefSeq release 62 now available
- NCBI's Eukaryotic Annotation Pipeline has now annotated the genomes for 100 different organisms!
- NCBI's 25th Anniversary and The Jim Gray eScience Award
- New SNP data available for several organisms!
- Update on PubMed Commons' comments in the early pilot phase
- NCBI News, November 2013 - NCBI NewsNCBI News, November 2013 - NCBI News
Your browsing activity is empty.
Activity recording is turned off.
See more...