

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
NCBI News [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 1991-2012.

April 26th NCBI Minute: Medical Genetics Summaries on the NCBI Bookshelf - a pharmacogenomics resource for clinicians
Wednesday, April 19, 2017
Next Wednesday, April 26, 2017, NCBI staff will introduce the Medical Genetics Summaries, a growing collection of reviews available on the NCBI Bookshelf. Each chapter of this book highlights the impact of genetic variations on response to drugs (pharmacogenomics). By the end of this NCBI Minute, you will be able to use the Medical Genetics Summaries to find information about a particular drug, including known impacts of genetic variation on drug response (efficacy, toxicity, side effects) and identify actionable information, including information about relevant genetic testing and how to interpret the test results in order to optimize therapy based on a patient's genotype.
Date and time: Wednesday, April 26, 2017 1:00 PM - 1:30 PM EDT
Register here.
After registering, you will receive a confirmation email with information about attending the webinar. After the live presentation, the webinar will be uploaded to the NCBI YouTube channel. Any related materials will be accessible on the Webinars and Courses page; you can also learn about future webinars on this page.
Maize (Zea mays) genome annotation release 101 is now available!
Wednesday, April 12, 2017
A new maize (Zea mays) genome annotation has been produced by the RefSeq eukaryotic genome annotation pipeline. In Annotation Release 101 a total of 47,446 genes were annotated, including 37,380 that code for proteins. This data is now available for download and can be explored in the Genome Data Viewer, with BLAST, and in the Gene database.
This annotation benefited from an improved assembly (B73 RefGen_v4), nearly 2 billion more RNA-Seq reads than the previous annotation, and information from over 750,000 PacBio Iso-Seq transcripts. As a result of the improved assembly and the increase in evidence used for gene prediction, about a third of the genes annotated are either new or substantially changed and 20% of the genes from the previous annotation (release 100) were dropped from the current one. In addition, manual curation of over 4000 genes was added by RefSeq Staff.
A full report on the maize (Zea mays) Annotation Release 101 annotation can be found here.
dbSNP FTP file format change planned for early 2018
Tuesday, April 11, 2017
In early 2018, dbSNP will no longer provide relational database table dumps on the FTP site or any general SQL support for future build releases. Instead, dbSNP FTP data will be made available as a cumulative file of RefSNP objects in JSON format. These files are now available, so users can begin migration and testing. Please see the dbSNP Alert README file for more details.
dbSNP's human build 150 has doubled the amount of RefSNP records!
Tuesday, April 11, 2017
dbSNP’s Human Build 150 includes a large number of new submissions from the Human Longevity, Inc. (HLI) and TopMed, increasing the total number of Human RefSNPs in the database from 154 to 324 million. TopMed has also provided new allele frequency data for 163 million RefSNPs.
Human Build 150 Notes:
HLI-submitted data were aligned with the human genome assembly GRCh38. Because dbSNP’s pipeline does not support mapping backward to previous assemblies, the rsIDs for these will not appear in the VCF files for GRCh37. We are investigating mechanisms to map these variants from the GRCh38 to GRCh37 assembly and will provide updates.
TopMed-submitted allele frequencies are available for both GRCh38.p7 and GRCh37.p13 in VCF format on the FTP site with the INFO tag 'TOPMED'.
Due to the unexpected increase in the volume of human data and limitations in our systems, dbSNP had to take two temporary actions for this release:
- The "dbSNP Build 150 (Homo sapiens Annotation Release 108) all data" annotation track for RefSeq genomic sequences will be limited to variants in the gene regions only. This only affect tracks displayed in the NCBI Sequence Viewer and does not impact reporting in dbSNP FTP files or on Reference SNP pages. We are investigating mechanisms to restore complete annotation across the genome and will provide updates.
- Entrez searching is currently only available for Human Build 150 and a limited number of organisms from previous builds - including mouse, rat, cow, and pig. We will provide updates over the next two weeks as we restore the search capability for other organisms.
For more information, see the dbSNP-Announce message.
NCBI researchers and collaborators discover novel group of giant viruses
Thursday, April 06, 2017
Nearly complete set of translation-related genes lends support to hypothesis that giant viruses evolved from smaller viruses
An international team of researchers, including NCBI’s Eugene Koonin and Natalya Yutin, has discovered a novel group of giant viruses (dubbed “Klosneuviruses”) with a more complete set of translation machinery genes than any virus that has been described to date. “This discovery significantly expands our understanding of viral evolution,” said Koonin. “These are the most ‘cell-like’ viruses ever identified. However, the computational analysis of the virus genomes shows that these viruses have not evolved from cells by reductive evolution but rather have evolved from smaller viruses, gradually acquiring genes from their hosts at different stages of their evolution.”
The research was published in the journal Science on April 6, 2017. In addition to biologists from NCBI, the authors include collaborators from the U.S. Department of Energy Joint Genome Institute (DOE JGI), the University of Vienna, and CalTech.
JGI researchers Frederik Schulz and Tanja Woyke unearthed Klosneuvirus while analyzing microcolony sequence data from a wastewater treatment plant sample in Klosterneuburg, Austria. "We expected nitrifier genome sequences in the microcolony sequence data,” Woyke said. “Finding a giant virus genome took the project into a completely new and unexpected, yet very exciting, direction." When Schulz noticed that several of the metagenomes were viral in origin, he and Woyke conducted analyses to determine their source. They found that the Klosneuvirus group came from a novel viral lineage affiliated with Mimiviruses, the first giant viruses discovered. A handful of other giant virus groups have been found since the discovery of Mimiviruses in 2003.
Giant viruses are characterized by disproportionately large genomes and virions that house viruses’ genetic material. They can encode several genes potentially involved in protein biosynthesis, a unique feature that has led to diverging hypotheses about their origin.Two evolutionary hypotheses have emerged. One posits that giant viruses evolved from an ancient cell, perhaps one from an extinct fourth domain of cellular life. Another — a scenario championed by Koonin — presents the idea that giant viruses descended from smaller viruses. The discovery of Klosneuvirus, Woyke said, supports the latter hypothesis. In this scenario, a smaller virus infected different eukaryote hosts and picked up genes from independent sources over long periods of time through piecemeal acquisition of translational machinery components.
“At first glance, the suite of “cellular” genes in Klosneuvirus seemed to have a common origin, but when we analyzed them in detail, we saw they came from different hosts,” Koonin said. “We could infer from the evolutionary trees we built that they have been acquired by the viruses piecemeal, at different stages in their evolution.” The Klosneuvirus genes contained aminoacyl-tRNA (transfer ribonucleic acid) enzymes with specificity for 19 out of 20 amino acids, along with more than 20 tRNAs and an array of translation factors and tRNA modifying enzymes—an unprecedented finding among any viruses, including the previously known giant viruses.
Schulz noted that while the metagenomic discovery of Klosneuviruses helped answer important evolutionary questions, the actual biological function of the translation system genes remains elusive—at least until these viruses are grown in the laboratory together with their hosts.
And Koonin believes there are more giant viruses waiting to be discovered in metagenomic data. “I’m quite confident that the current record of the genome size of giant viruses will be broken,” he said. “We are going to see the real Goliaths of the giant virus world.”
— Many thanks to JGI for their assistance in preparing this news feature.
Image credit: Ella Maru studio
April 19th NCBI Minute: Magic-BLAST, NCBI's next-gen sequence alignment program
Wednesday, April 05, 2017
In two weeks, NCBI staff will introduce you to Magic-BLAST, NCBI's next-gen sequence aligner. You will learn to use magic-BLAST to align next-gen RNA and DNA sequencing runs to genomic and transcript sequences and to understand the options available for magic-BLAST
Date and time: Wednesday, April 19, 2017 12:00 PM - 12:30 PM EDT
After registering, you will receive a confirmation email with information about attending the webinar. After the live presentation, the webinar will be uploaded to the NCBI YouTube channel. Any related materials will be accessible from the Webinars and Courses page; you can also learn about future webinars on this page.
Six functional prototypes available from the March NCBI hackathon
Wednesday, April 05, 2017
At the March 2017 NCBI Genomics Hackathon, participants developed six functional software prototypes, several of which are still under active development. Software is available from the NCBI-Hackathons GitHub site.
- Squidstream provides naming consistency by converting sequence feature IDs in entire files (bed, gff3, wig, etc.) to the desired ID format using a single command.
- ga4gh-ncbi-api is a method that links NCBI’s API and the GA4GH (Global Alliance for Genomics and Health) API, and generates a searchable list of genome datasets from NCBI.
- Graph_Extraction provides code to implement a simple graph genome browser.
- Sidearm searches the SRA database for viruses using the NCBI magicBLAST tool.
- Scan2CNV is a commandline tool that generates copy number variation (CNV) calls from raw SNP array data.
- Single Cell Reproducible Epigenomics Workflow (SCREW) is a single-cell whole-genome bisulfite sequencing (SC-WGBS) pipeline and docker image for performing standard single-cell DNA methylation analyses.
Eight new eukaryotic genome annotations added to RefSeq
Tuesday, April 04, 2017
In the past month, the NCBI Eukaryotic Genome Annotation Pipeline has released new annotations in RefSeq for the following organisms:
- Zea mays (maize)
- Labrus bergylta (ballan wrasse)
- Monopterus albus (swamp eel)
- Corvus cornix cornix (hooded crow)
- Prunus persica (peach)
- Rhincodon typus (whale shark)
- Oncorhynchus kisutch (coho salmon)
- Pseudomyrmex gracilis (ant)
See more details on the Eukaryotic RefSeq Genome Annotation Status page.

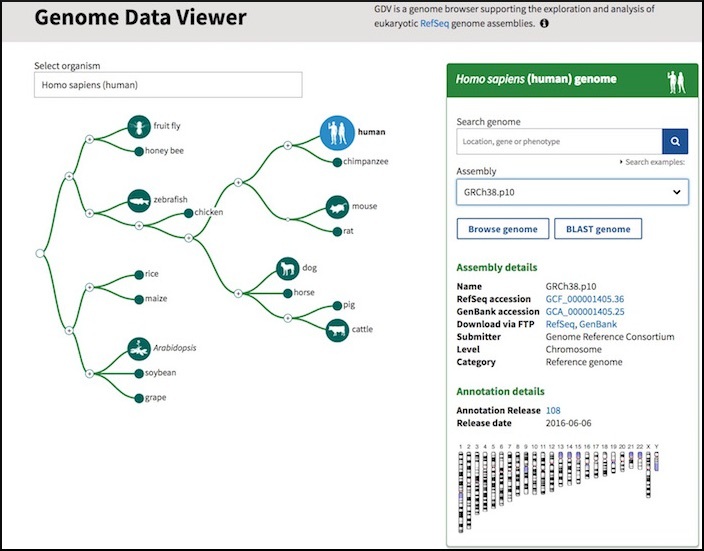
New Genome Data Viewer access page
Monday, April 03, 2017
NCBI is pleased to offer a direct entry point to the NCBI Genome Data Viewer (GDV) that supports the exploration, visualization and analysis of eukaryotic RefSeq genome assemblies. The new GDV homepage includes an interactive interface for a quick overview of supported organisms, specific genome searches plus inter-connectivity to Assembly and RefSeq annotation resources. About 100 genome assemblies are now ready for GDV exploration with more on the way. Stay tuned!
- April 26th NCBI Minute: Medical Genetics Summaries on the NCBI Bookshelf - a pharmacogenomics resource for clinicians
- Maize (Zea mays) genome annotation release 101 is now available!
- dbSNP FTP file format change planned for early 2018
- dbSNP's human build 150 has doubled the amount of RefSNP records!
- NCBI researchers and collaborators discover novel group of giant viruses
- April 19th NCBI Minute: Magic-BLAST, NCBI's next-gen sequence alignment program
- Six functional prototypes available from the March NCBI hackathon
- Eight new eukaryotic genome annotations added to RefSeq
- New Genome Data Viewer access page
- NCBI News, April 2017 - NCBI NewsNCBI News, April 2017 - NCBI News
Your browsing activity is empty.
Activity recording is turned off.
See more...