This chapter sets out in detail the methods used to review the evidence and to generate the recommendations that are presented in subsequent chapters. This guidance was developed in accordance with the methods outlined in the NICE guidelines manual 2012.125
3.1. Developing the review questions and outcomes
Review questions were developed in a PICO framework (patient, intervention, comparison and outcome) for intervention reviews; in a framework of population, index tests, reference standard and target condition for reviews of diagnostic test accuracy; and using population, presence or absence of factors under investigation (for example prognostic factors) and outcomes for prognostic reviews. The experience of information provision for people with suspected or confirmed drug allergies was reviewed using qualitative information to capture preferences and perceptions (including factors which improve or act as a barrier to optimal care).
This use of a framework guided the literature searching process, critical appraisal and synthesis of evidence, and facilitated the development of recommendations by the GDG. The review questions were drafted by the NCGC technical team and refined and validated by the GDG. The questions were based on the key clinical areas identified in the scope (Appendix A).
A total of 11 review questions were identified.
Full literature searches, critical appraisals and evidence reviews were completed for all the specified review questions.
Table 1
Review questions.
3.2. Searching for evidence
3.2.1. Clinical literature search
Systematic literature searches were undertaken to identify all published clinical evidence relevant to the review questions. Searches were undertaken according to the parameters stipulated within the NICE guidelines manual 2012.125 Databases were searched using relevant medical subject headings, free-text terms and study-type filters where appropriate. Studies published in languages other than English were not reviewed. Where possible, searches were restricted to articles published in English. All searches were conducted in MEDLINE, Embase, and The Cochrane Library. In additional Cinahl was used for the information and support review. All searches were updated on 10 January 2014. No papers published after this date were considered.
Search strategies were quality assured by cross-checking reference lists of highly relevant papers, analysing search strategies in other systematic reviews, and asking GDG members to highlight any additional studies. The questions, the study types applied, the databases searched and the years covered can be found in Appendix G.
The titles and abstracts of records retrieved by the searches were sifted for relevance, with potentially significant publications obtained in full text. These were assessed against the inclusion criteria.
During the scoping stage, a search was conducted for guidelines and reports on the websites listed below from organisations relevant to the topic. Searching for unpublished literature was not undertaken. All references sent by stakeholders were considered.
- Guidelines International Network database (http://www.g-i-n.net/)
- National Guideline Clearing House (http://www.guideline.gov/)
- National Institute for Health and Care Excellence (NICE) (http://www.nice.org.uk/)
- National Institutes of Health Consensus Development Program (http://consensus.nih.gov/)
- NHS Evidence Search (http://www.evidence.nhs.uk/)
- British Society for Allergy & Clinical Immunology (BSACI) (http://www.bsaci.org/)
- American Academy of Allergy, Asthma & Immunology (AAAAI) (http://www.aaaai.org/home.aspx).
3.2.2. Health economic literature search
Systematic literature searches were also undertaken to identify health economic evidence within published literature relevant to the review questions. The evidence was identified by conducting a broad search relating to drug allergy in the NHS Economic Evaluation Database (NHS EED), the Health Technology Assessment database (HTA) and the Health Economic Evaluations Database (HEED) with no date restrictions. Additionally, the search was run on MEDLINE and Embase using a specific economic filter, from 2011, to ensure recent publications that had not yet been indexed by the economic databases were identified. Studies published in languages other than English were not reviewed. For databases, where it was possible, searches were restricted to articles published in English.
The health economic search strategies are included in Appendix G. All searches were updated on 15 January 2014. No papers published after this date were considered.
3.3. Evidence of effectiveness
The evidence was reviewed following the steps shown schematically in Figure 1:

Figure 1
Step-by-step process of review of evidence in the guideline.
- Potentially relevant studies were identified for each review question from the relevant search results by reviewing titles and abstracts. Full papers were then obtained.
- Full papers were reviewed against pre-specified inclusion and exclusion criteria to identify studies that addressed the review question in the appropriate population (review protocols are included in Appendix C).
- Key information was extracted on the study's methods, PICO factors and results. These were presented in summary tables (in each review chapter) and evidence tables (in Appendix H).
- Summaries of evidence were generated by outcome (included in the relevant review chapters) and were presented in GDG meetings:
- Randomised studies: data were meta-analysed where appropriate and reported in GRADE profiles (for intervention reviews).
- Observational studies: data were presented as a range of values in GRADE profiles.
- Prognostic studies: data were presented as a range of values, usually in terms of the relative effect as reported by the authors.
- Diagnostic studies were presented as measures of diagnostic test accuracy (sensitivity, specificity, positive and negative predictive value). Coupled values of sensitivity and specificity were summarised in paired (sensitivity and specificity side by side) forest plots to allow visual comparison between different index tests and to investigate heterogeneity more effectively (given data were reported at the same thresholds).
- Qualitative studies: each study was summarised in a table where possible, otherwise presented in a narrative.
A 20% sample of each of the above stages of the reviewing process was quality assured by a second reviewer to eliminate any potential of reviewer bias or error.
3.3.1. Inclusion and exclusion criteria
The inclusion and exclusion of studies was based on the review protocols, which can be found in Appendix C. Excluded studies by review question (with the reasons for their exclusion) are listed in Appendix K. The GDG was consulted about any uncertainty regarding inclusion or exclusion.
The guideline population was defined to be people with suspected or confirmed drug allergies. For some review questions, the review population was defined by the drug or drug class the person was allergic to (for example beta-lactam antibiotics, non-steroidal anti-inflammatories, local anaesthetics or general anaesthetics in review questions 8 to 11).
In the diagnostic chapter serum IgE testing was reviewed for a list of drugs that was prioritised by the GDG: amoxicillin, ampicillin, cefaclor, chlorhexidine, morphine, penicillin G, penicillin V, and suxamethonium.
The diagnostic serum tryptase review was restricted to patients with signs and symptoms of anaphylaxis.
Even though the prognostic review (to examine if there were certain characteristics of people with an allergy to NSAIDs who could take selective COX-2 inhibitors) had identified specific characteristics as prognostic factors, studies that were not designed to directly address these factors were not excluded. Studies that investigated the safety of taking selective COX-2 inhibitors for people with an allergy to NSAIDs more generally were included as indirect evidence. These studies were then divided by the study population (people with asthma or people with cutaneous reactions) to address the prognostic aspect of the question. For details of the approach to this review please refer to Chapter 11.
Randomised trials, non-randomised trials, and observational studies (including diagnostic or prognostic studies) were included in the evidence reviews as appropriate.
Conference abstracts were not automatically excluded from the review but were initially assessed against the inclusion criteria and then further processed only if no other full publication was available for that review question, in which case the authors of the selected abstracts were contacted for further information. None of the reviews in this guideline included conference abstracts as part of the evidence.
Literature reviews, posters, letters, editorials, comment articles, unpublished studies and studies not in English were excluded.
The review protocols are presented in Appendix C.
3.3.2. Methods of combining clinical studies
3.3.2.1. Data synthesis for intervention reviews
Where possible, meta-analyses were conducted to combine the results of studies for each review question using Cochrane Review Manager (RevMan5) software. Fixed-effects (Mantel-Haenszel) techniques were used to calculate risk ratios (relative risk) for the binary outcomes, such as number of patients being treated with alternative beta-lactam antibiotics, or number of patients with medication errors.
For continuous outcomes, measures of central tendency (mean) and variation (standard deviation) were required for meta-analysis. Data for continuous outcomes (such as prescription errors) were analysed using an inverse variance method for pooling weighted mean differences and, where the studies had different scales, standardised mean differences were used. A generic inverse variance option in RevMan5 was used if any studies reported solely the summary statistics and 95% confidence interval (95% CI) or standard error; this included any hazard ratios reported. However, in cases where standard deviations were not reported per intervention group, the standard error (SE) for the mean difference was calculated from other reported statistics (p values or 95% CIs); meta-analysis was then undertaken for the mean difference and SE using the generic inverse variance method in RevMan5. When the only evidence was based on studies that summarised results by presenting medians (and interquartile ranges), or only p values were given, this information was assessed in terms of the study's sample size and was included in the GRADE tables without calculating the relative or absolute effects. Consequently, aspects of quality assessment such as imprecision of effect could not be assessed for evidence of this type.
Statistical heterogeneity was assessed by visually examining the forest plots, and by considering the chi-squared test for significance at p<0.1 or an I-squared inconsistency statistic (with an I-squared value of more than 50% indicating considerable heterogeneity). Where considerable heterogeneity was present, we carried out predefined subgroup analyses for type of drug allergy and age group (children or adults).
The means and standard deviations of continuous outcomes were required for meta-analysis. However, in cases where standard deviations were not reported, the standard error was calculated if the p values or 95% CIs were reported and meta-analysis was undertaken with the mean and standard error using the generic inverse variance method in RevMan5. Where p values were reported as ‘less than’, a conservative approach was undertaken. For example, if p value was reported as ‘p≤0.001’, the calculations for standard deviations will be based on a p value of 0.001.
For interpretation of the binary outcome results, differences in the absolute event rate were calculated using the GRADEpro software, for the median event rate across the control arms of the individual studies in the meta-analysis. Absolute risk differences were presented in the GRADE profiles and in clinical summary of findings tables, for discussion with the GDG.
For binary outcomes, absolute event rates were also calculated using the GRADEpro software using event rate in the control arm of the pooled results.
3.3.2.2. Data synthesis for prognostic factor reviews
Odds ratios (ORs), risk ratios (RRs) or hazard ratios (HRs), with their 95% confidence intervals (95% CIs) for the effect of the pre-specified prognostic factors were extracted from the papers when reported. For the purpose of the review question on tolerance of selective COX-2 inhibitors, factors that indicated whether the drug was safe to prescribe regardless of prognostic factors were also noted, such as the type of allergic reaction and the rate of severe reactions in response to the selective COX-2 inhibitor.
3.3.2.3. Data synthesis for diagnostic test accuracy reviews
Data and outcomes
For the reviews of diagnostic test accuracy, a positive result on the index test was found if the patient had values of the measured quantity above a threshold value, and different thresholds could be used. Diagnostic test accuracy measures used in the analysis were: sensitivity, specificity, positive and negative predictive value. The threshold of a diagnostic test is defined as the value at which the test can best differentiate between those with and without the target condition (for instance different thresholds were used in the serum tryptase review) and, in practice, it varies amongst studies. For this guideline, sensitivity and specificity were considered equally important. A high sensitivity (true positives) of a test can pick up the majority of the correct cases with drug allergy; conversely, a high specificity (true negatives) can correctly exclude people without drug allergy.
Data synthesis
Coupled forest plots of sensitivity and specificity with their 95% CIs across studies (at various thresholds) were produced for each test, using RevMan5. In order to do this, 2×2 tables (the number of true positives, false positives, true negatives and false negatives) were directly taken from the study if given, or else were derived from raw data or calculated from the set of test accuracy statistics.
Heterogeneity or inconsistency amongst studies was visually inspected in the forest plots where appropriate (only when there were similar thresholds). A diagnostic meta-analysis was not carried out because studies were not homogenous enough to assume a single underlying level of sensitivity and specificity (due to differences in population, type of index test or reference standard).
3.3.2.4. Data synthesis for qualitative study review
Where possible a meta-synthesis was conducted to combine qualitative study results. The main aim of the synthesis of qualitative data was a description of the main topics that may influence the experience of care of the person with suspected or confirmed drug allergy or their parents or carers, rather than build new theories or reconceptualise the topic under review. Whenever studies identified a qualitative theme, this was extracted and the main characteristics were summarised. When all themes were extracted from studies, common concepts were categorised and tabulated. This included information on how many studies had identified this theme. A frequently identified theme may indicate an important issue for the review, but frequency of theme is not the only indicator of importance. Study type and population in qualitative research can differ widely meaning that themes that may only be identified by one or a few studies can provide important new information. Therefore for the purpose of the qualitative review in this guideline the categorisation of themes was exhaustive, that is all themes were accounted for in the synthesis. The GDG could then draw conclusions on the relative merits of each of the themes and how they may help in forming recommendations.
3.3.2.5. Data synthesis for the algorithm and probability score review
The aim of this review was to summarise evidence on issues that clinicians need to consider when assessing a person with a suspected drug allergy and the signs and symptoms that the person would present with. Assessments should be suitable for the primary care setting. It was decided that this topic would be best addressed with a review of already published assessment methods (that is, algorithms and probability scores) because of the multitude of individual features that may indicate a potential drug allergy. After a top-level search on this topic a published systematic review was identified (Agbabiaka 20083). This review was edited (studies restricted to adverse drug events without drug allergies were excluded) and updated. For a full description of this specific data synthesis approach please see Chapter 6.
3.3.2.6. Data synthesis for the documentation review
The aim of this review was to summarise evidence on the effectiveness of documentation strategies in preventing people with suspected or confirmed allergies receiving the drug they are allergic to. Study types considered for this review were randomised trials, and systematic reviews. Prospective and retrospective cohort studies, before and after studies, case series, surveys and qualitative studies were also considered, with the caveat that if a lot of evidence was identified for a particular documentation intervention then only the higher-level evidence be included in the review.
Due to the multitude of populations, study designs, interventions and reported outcomes an exception was made to the usual effectiveness reviews described above. The following approach was used:
- Evidence was classified according to the broad documentation category (for example, computerised systems or structured charts).
- Features of different documentation categories were then extracted.
- Outcomes (such as prescribing errors or alerts that were overwritten) were summarised and where possible related to the features of the documentation strategy.
- Study quality was assessed individually and then by the majority of evidence for a particular intervention and outcome.
- Overall quality was then assessed by documentation category.
Further details of this approach are described in Chapter 9.
3.3.3. Type of studies
For most intervention reviews in this guideline, parallel randomised controlled trials (RCTs) were included because they are considered the most robust type of study design that could produce an unbiased estimate of the intervention effects. If the GDG believed RCT data were not appropriate or there was limited evidence from RCTs, well-conducted non-randomised studies were included. Please refer to Appendix C for full details on the study design of studies selected for each review question. For example in the questions on referral to specialist drug allergy services it was decided to include non-randomised trials since randomisation might not always be possible or appropriate.
For the diagnostic reviews and the algorithm and probability score review, cross-sectional and retrospective studies were included. For prognostic reviews, prospective and retrospective cohort studies were included.
Where data from observational studies were included, the GDG decided that the results for each outcome should be presented separately for each study and meta-analysis was not conducted.
3.3.4. Appraising the quality of evidence by outcomes
The evidence for outcomes from the included RCTs and, where appropriate, observational studies were evaluated and presented using an adaptation of the ‘Grading of Recommendations Assessment, Development and Evaluation (GRADE) toolbox’ developed by the international GRADE working group (http://www.gradeworkinggroup.org/). The software developed by the GRADE working group (GRADEpro) was used to assess the quality of each outcome, taking into account individual study quality factors and the meta-analysis results. Results were presented in GRADE profiles (‘GRADE tables’), which consist of 2 sections: the ‘Clinical evidence profile’ table includes details of the quality assessment while the ‘Clinical evidence summary of findings’ table includes pooled outcome data, where appropriate, an absolute measure of intervention effect and the summary of quality of evidence for that outcome. In this table, the columns for intervention and control indicate summary measures and measures of dispersion (such as mean and standard deviation or median and range) for continuous outcomes and frequency of events (n/N: the sum across studies of the number of patients with events divided by sum of the number of completers) for binary outcomes. Reporting or publication bias was only taken into consideration in the quality assessment and included in the ‘Clinical evidence profile’ table if it was apparent.
The evidence for each outcome was examined separately for the quality elements listed and defined in Table 2. Each element was graded using the quality levels listed in Table 3. The main criteria considered in the rating of these elements are discussed below (see Section 3.3.5 Grading of evidence). Footnotes were used to describe reasons for grading a quality element as having serious or very serious problems. The ratings for each component were summed to obtain an overall assessment for each outcome (Table 4).
Table 2
Description of the elements in GRADE used to assess the quality of intervention and diagnostic studies.
Table 3
Levels of quality elements in GRADE.
Table 4
Overall quality of outcome evidence in GRADE.
The GRADE toolbox is currently designed only for randomised trials and observational studies but we adapted the quality assessment elements and outcome presentation for diagnostic accuracy studies.
3.3.5. Grading the quality of clinical evidence
After results were pooled, the overall quality of evidence for each outcome was considered. The following procedure was adopted when using GRADE:
- 3.
A quality rating was assigned, based on the study design. RCTs start as High, observational studies as Low, and uncontrolled case series as Low or Very low.
- 4.
The rating was then downgraded for the specified criteria: risk of bias (study limitations), inconsistency, indirectness, imprecision and publication bias. These criteria are detailed below. Evidence from observational studies (which had not previously been downgraded) was upgraded if there was: a large magnitude of effect, a dose–response gradient, and if all plausible confounding would reduce a demonstrated effect or suggest a spurious effect when results showed no effect. Each quality element considered to have ‘serious’ or ‘very serious’ risk of bias was rated down by 1 or 2 points respectively.
- 5.
The downgraded or upgraded marks were then summed and the overall quality rating was revised. For example, all RCTs started as High and the overall quality became Moderate, Low or Very low if 1, 2 or 3 points were deducted respectively.
- 6.
The reasons or criteria used for downgrading were specified in the footnotes.
The details of the criteria used for each of the main quality element are discussed further in the following Sections 3.3.6 to 3.3.9.
3.3.6. Risk of bias
Bias can be defined as anything that causes a consistent deviation from the truth. Bias can be perceived as a systematic error, for example, if a study was to be carried out several times and there was a consistently wrong answer, the results would be inaccurate.
The risk of bias for a given study and outcome is associated with the risk of over- or underestimation of the true effect.
The risks of bias are listed in Table 5.
Table 5
Risk of bias in randomised controlled trials.
A study with a poor methodological design does not automatically imply high risk of bias; the bias is considered individually for each outcome and it is assessed whether this poor design will impact on the estimation of the intervention effect.
3.3.6.1. Diagnostic studies
For diagnostic accuracy studies, the Quality Assessment of Diagnostic Accuracy Studies version 2 (QUADAS-2) checklist was used (see Appendix F of The guidelines manual (2012)125). Risk of bias and applicability in primary diagnostic accuracy studies in QUADAS-2 consists of 4 domains (see Figure 2):
Figure 2
Summary of QUADAS-2 checklist.
- patient selection
- index test
- reference standard
- flow and timing.
Optional domain, multiple test accuracy is applicable when a single study examined more than 1 diagnostic test (head-to-head comparison between 2 or more index tests reported within the same study). This optional domain contains 3 questions relating to risk of bias:
- Did all patients undergo all index tests or were the index tests appropriately randomised amongst the patients?
- Were index tests conducted within a short time interval?
- Are index test results unaffected when undertaken together on the same patient?
3.3.6.2. Prognostic studies
For prognostic studies, quality was assessed using the checklist for prognostic studies (Appendix I in The guidelines manual (2012)125). The quality rating (Low, High, Unclear) was derived by assessing the risk of bias across 6 domains: selection bias, attrition bias, prognostic factor bias, outcome measurement bias, control for confounders and appropriate statistical analysis, with the last 4 domains being assessed for each outcome. A summary table on the quality of prognostic studies is presented at the beginning of each review to summarise the risk of bias across the 5 domains. More details about the quality assessment for prognostic studies are shown below:
- The study sample represents the population of interest with regard to key characteristics
- Missing data are unrelated to key characteristics, sufficient to limit potential bias – reasons for missing data are adequately described.
- The prognostic factor of interest is adequately measured in study participants.
- The outcome of interest is adequately measured in study participants.
- Important potential confounders are accounted for appropriately.
- The statistical analysis is appropriate for the design of the study, limiting potential for the presentation of valid results.
Many of the studies in the prognostic review were safety studies, that is, they did not directly investigate particular factor and in these cases the checklist for non-randomised studies was used.
3.3.6.3. Qualitative studies
For qualitative studies, quality was assessed using the checklist for qualitative studies (Appendix I in The guidelines manual (2012)125). The quality rating (Low, High, Unclear) was derived by assessing the risk of bias across 6 domains:
- theoretical approach
- study design
- data collection
- validity
- analysis
- ethics.
3.3.6.4. Algorithm and probability score studies
For these studies none of the checklists adequately addressed the specific quality criteria deemed important by the GDG. A checklist was therefore designed for these studies to assess risk of bias across 9 criteria:
These criteria were based on factors that were considered in the narrative assessment of the algorithms within the systematic review3 included in the chapter (see 6.2.1):
- Design of the tool:How the tool was designed in a systematic way (that is, using a statistical method or by way of literature review)
- Factors that are considered:Whether or not a sufficient number of features were considered
- Applicability to clinical practice (primary care):The aim of the review was to find an assessment that could be carried out in general practice and was therefore applicable to current practice.
- Definition of condition:Whether the tool was based on a clear definition of the condition for which it was going to be used.
- Number of evaluators or assessors:Whether in the design of the tool separate independent evaluators were used and their assessments were analysed for consistency.
- Prior probabilities:Whether the group that was assessed was generalisable to the general population of people with drug allergies or whether only high risk participants were assessed by the tool.
- Validation in independent studies:Whether this test has been further used as a reference standard in other test comparisons
- Confounders or alternative conditions:Whether plausible alternative conditions or factors that may affect the result of the algorithm were considered
- Ease of interpretation:Whether the test could be easily and quickly scored and also whether the result or ‘score’ could be easily interpreted in primary care.
This quality checklist was used by 2 reviewers independently and differences in assessments were discussed and agreed.
3.3.7. Inconsistency
Inconsistency refers to an unexplained heterogeneity of results. When estimates of the treatment effect across studies differ widely (that is, there is heterogeneity or variability in results), this suggests true differences in underlying treatment effect.
Heterogeneity in meta-analyses was examined and sensitivity and subgroup analyses performed as pre-specified in the protocols (Appendix C).
When heterogeneity exists (chi-squared p<0.1, I-squared inconsistency statistic of >50%, or evidence from examining forest plots), but no plausible explanation can be found (for example, duration of intervention or different follow-up periods), the quality of evidence was downgraded by 1 or 2 levels, depending on the extent of uncertainty to the results contributed by the inconsistency in the results. In addition to the I-squared and chi-squared values, the decision for downgrading was also dependent on factors such as whether the intervention is associated with benefit in all other outcomes or whether the uncertainty about the magnitude of benefit (or harm) of the outcome showing heterogeneity would influence the overall judgment about net benefit or harm (across all outcomes).
3.3.8. Indirectness
Directness refers to the extent to which the populations, intervention, comparisons and outcome measures are similar to those defined in the inclusion criteria for the reviews. Indirectness is important when these differences are expected to contribute to a difference in effect size, or may affect the balance of harms and benefits considered for an intervention.
3.3.9. Imprecision
Imprecision in guidelines concerns whether the uncertainty (confidence interval) around the effect estimate means that it is not clear whether there is a clinically important difference between interventions or not. Therefore, imprecision differs from the other aspects of evidence quality, in that it is not really concerned with whether the point estimate is accurate or correct (has internal or external validity) instead it is concerned with the uncertainty about what the point estimate is. This uncertainty is reflected in the width of the confidence interval.
The 95% confidence interval (95% CI) is defined as the range of values that contain the population value with 95% probability. The larger the trial, the smaller the 95% CI and the more certain the effect estimate.
Imprecision in the evidence reviews was assessed by considering whether the width of the 95% CI of the effect estimate is relevant to decision-making, considering each outcome in isolation. Figure 3 considers a positive outcome for the comparison of treatment A versus B. Three decision-making zones can be identified, bounded by the thresholds for clinical importance (minimal important difference – MID) for benefit and for harm. The MID for harm for a positive outcome means the threshold at which drug A is less effective than drug B by an amount that is clinically important to patients (favours B).
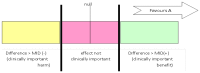
Figure 3
Illustration of precise and imprecise outcomes based on the confidence interval of outcomes in a forest plot.
When the confidence interval of the effect estimate is wholly contained in one of the 3 zones (for example, clinically important benefit), we are not uncertain about the size and direction of effect (whether there is a clinically important benefit, or the effect is not clinically important, or there is a clinically important harm), so there is no imprecision.
When a wide confidence interval lies partly in each of 2 zones, it is uncertain in which zone the true value of effect estimate lies, and therefore there is uncertainty over which decision to make (based on this outcome alone). The confidence interval is consistent with 2 decisions and so this is considered to be imprecise in the GRADE analysis and the evidence is downgraded by 1 level (‘serious imprecision’).
If the confidence interval of the effect estimate crosses into 3 zones, this is considered to be very imprecise evidence because the confidence interval is consistent with 3 clinical decisions and there is a considerable lack of confidence in the results. The evidence is therefore downgraded by 2 levels in the GRADE analysis (‘very serious imprecision’).
Implicitly, assessing whether the confidence interval is in, or partially in, a clinically important zone, requires the GDG to estimate an MID or to say whether they would make different decisions for the 2 confidence limits.
The literature was searched for established MIDs for the selected outcomes in the evidence reviews. In addition, the GDG was asked whether they were aware of any acceptable MIDs in the clinical community. Finally, the GDG considered it clinically acceptable to use the GRADE default MID to assess imprecision: a 25% relative risk reduction or relative risk increase was used, which corresponds to clinically important thresholds for a risk ratio of 0.75 and 1.25 respectively. This default MID was used for all the outcomes in the interventions evidence reviews.
3.3.10. Evidence statements
Evidence statements are summary statements that are presented after the GRADE profiles, summarising the key features of the clinical effectiveness evidence presented. The wording of the evidence statements reflects the certainty or uncertainty in the estimate of effect. The evidence statements are presented by outcome and encompass the following key features of the evidence:
- the number of studies and the number of participants for a particular outcome
- a brief description of the participants
- an indication of the direction of effect (if one treatment is beneficial or harmful compared to the other, or whether there is no difference between the 2 tested treatments)
- a description of the overall quality of evidence (GRADE overall quality).
3.4. Evidence of cost effectiveness
The GDG is required to make decisions based on the best available evidence of both clinical and cost effectiveness. Guideline recommendations should be based on the expected costs of the different options in relation to their expected health benefits (that is, their ‘cost effectiveness’) rather than the total implementation cost.125 Thus, if the evidence suggests that a strategy provides significant health benefits at an acceptable cost per patient treated, it should be recommended even if it would be expensive to implement across the whole population.
Evidence on cost effectiveness related to the key clinical issues being addressed in the guideline was sought. The health economists:
- Undertook a systematic review of the published economic literature.
- Undertook new cost-effectiveness analysis in priority areas.
3.4.1. Literature review
The health economist:
- Identified potentially relevant studies for each review question from the economic search results by reviewing titles and abstracts. Full papers were then obtained.
- Reviewed full papers against pre-specified inclusion and exclusion criteria to identify relevant studies (see below for details).
- Critically appraised relevant studies using the economic evaluations checklist as specified in The guidelines manual (2012).125
- Extracted key information about the studies' methods and results into evidence tables (included in Appendix I).
- Generated summaries of the evidence in NICE economic evidence profiles (included in the relevant chapter for each review question) – see below for details.
3.4.1.1. Inclusion and exclusion criteria
Full economic evaluations (studies comparing costs and health consequences of alternative courses of action: cost–utility, cost-effectiveness, cost–benefit and cost–consequence analyses) and comparative costing studies that addressed the review question in the relevant population were considered potentially includable as economic evidence.
Studies that only reported cost per hospital (not per patient), or only reported average cost effectiveness without disaggregated costs and effects, were excluded. Literature reviews, abstracts, posters, letters, editorials, comment articles, unpublished studies and studies not in English were excluded. No economic evaluations were identified that related to a review question and satisfied these criteria. Therefore, no economic evaluations were included in this guideline.
Remaining studies would have been prioritised for inclusion based on their relative applicability to the development of this guideline and the study limitations. For example, if a high quality, directly applicable UK analysis had been available, then other less relevant studies may not have been included. However, no exclusions occurred on this basis in this guideline, as all studies had already been excluded on the grounds of not being a full economic evaluation or not relating to any of the review questions, and so no economic evaluations were listed in the excluded economic studies appendix (Appendix L).When no relevant economic studies were found from the economic literature review, relevant UK NHS unit costs related to the compared interventions were presented to the GDG to inform the possible economic implications of the recommendations.
3.4.2. Undertaking new health economic analysis
As well as reviewing the published economic literature for each review question, as described above, new economic analysis was undertaken by the health economists in selected areas. Priority areas for new health economic analysis were agreed by the GDG after formation of the review questions and consideration of the available health economic evidence.
The GDG identified referral to specialist drug allergy services or alternative management strategies within primary care for patients who are not referred as the highest priority area for original economic analysis. The GDG believed that economic modelling in this area would be informative if feasible, but concluded that modelling was unfortunately not feasible as information was not available on the relative effectiveness of referral or non-specialist management on outcomes such as the number of future allergic reactions or the number of occasions alternative drugs are used. This was due both to the fact that as specialist management is outside the scope of this guideline the referral pathway is undefined, and to the lack of applicable published economic research on the areas that are within the scope. Therefore any model would necessarily have to be built largely upon estimates and assumptions. In particular, sufficient data were not available to allow modelling of different subgroups, which would be necessary to identify which individuals should or should not be referred to specialist drug allergy services.
Instead of conducting a full economic evaluation, 4 cost-effectiveness scenarios were constructed for the case of suspected allergy to beta-lactam antibiotics. These calculated the potential costs of both referral to specialist drug allergy services and of non-specialist management for multiple frequencies of future need for antibiotics. They presented the magnitude of difference in quality of life (measured in quality-adjusted life years [QALYs] or life days [QALDs]) which referral would need to be expected to yield for it to be cost effective compared to non-specialist management. The GDG used these scenarios to inform their recommendations regarding which people should and should not be referred to specialist drug allergy services.
The following general principles were adhered to in developing the cost-effectiveness scenarios:
- Methods were consistent with the NICE reference case.126
- The GDG was involved in the design of the scenarios, selection of conditions and drugs examined and interpretation of the results.
- Costs were based on routine NHS data sources.
- Inputs and assumptions were reported fully and transparently, and their limitations were discussed.
Full methods for the cost-effectiveness scenarios for referral to specialist drug allergy services are described in Chapter 12.
3.4.3. Cost-effectiveness criteria
NICE's report ‘Social value judgements: principles for the development of NICE guidance’ sets out the principles that GDGs should consider when judging whether an intervention offers good value for money.123 In general, an intervention was considered to be cost effective if either of the following criteria applied (given that the estimate was considered plausible):
- the intervention dominated other relevant strategies (that is, it was both less costly in terms of resource use and more clinically effective compared with all the other relevant alternative strategies), or
- the intervention cost less than £20,000 per QALY gained compared with the next best strategy.
If the GDG recommended an intervention that was estimated to cost more than £20,000 per QALY gained, or did not recommend one that was estimated to cost less than £20,000 per QALY gained, the reasons for this decision are discussed explicitly in the ‘Recommendations and link to evidence’ section of the relevant chapter, with reference to issues regarding the plausibility of the estimate or to the factors set out in ‘Social value judgements: principles for the development of NICE guidance’.123
When QALYs or life years gained are not used in an analysis, results are difficult to interpret unless one strategy dominates the others with respect to every relevant health outcome and cost.
3.4.4. In the absence of economic evidence
When no relevant published studies were found, and a new analysis was not prioritised, the GDG made a qualitative judgement about cost effectiveness by considering expected differences in resource use between options and relevant UK NHS unit costs, alongside the results of the clinical review of effectiveness evidence.
3.5. Developing recommendations
Over the course of the guideline development process, the GDG was presented with:
- Evidence tables of the clinical and economic evidence reviewed from the literature. All evidence tables are in Appendices H and I.
- Summaries of clinical and economic evidence and quality (as presented in Chapters 6-12).
- Forest plots (Appendix J).
Recommendations were drafted on the basis of the GDG's interpretation of the available evidence, taking into account the balance of benefits, harms and costs between different courses of action. Firstly, the net benefit over harm (clinical effectiveness) was considered, focusing on the critical outcomes. When this was done informally, the GDG took into account the clinical benefits and harms when one intervention was compared with another. The assessment of net benefit was moderated by the importance placed on the outcomes (the GDG's values and preferences), and the confidence the GDG had in the evidence (evidence quality). Secondly, whether the net benefit justified any differences in costs was assessed.
When clinical and economic evidence was of poor quality, conflicting or absent, the GDG drafted recommendations based on their expert opinion. The considerations for making consensus-based recommendations include the balance between potential harms and benefits, the economic costs compared to the economic benefits, current practices, recommendations made in other relevant guidelines, patient preferences and equality issues. The consensus recommendations were agreed through discussions in the GDG. The GDG also considered whether the uncertainty was sufficient to justify delaying making a recommendation to await further research, taking into account the potential harm of failing to make a clear recommendation (see Section 3.5.1 below).
The GDG considered the ‘strength’ of recommendations. This takes into account the quality of the evidence but is conceptually different. Some recommendations are ‘strong’ in that the GDG believes that the vast majority of healthcare and other professionals and patients would choose a particular intervention if they considered the evidence in the same way that the GDG has. This is generally the case if the benefits clearly outweigh the harms for most people and the intervention is likely to be cost effective. However, there is often a closer balance between benefits and harms, and some patients would not choose an intervention whereas others would. This may happen, for example, if some patients are particularly averse to some side effect and others are not. In these circumstances the recommendation is generally weaker, although it may be possible to make stronger recommendations about specific groups of patients.
The GDG focused on the following factors in agreeing the wording of the recommendations:
- The actions health professionals need to take.
- The information readers need to know.
- The strength of the recommendation (for example the word ‘offer’ was used for strong recommendations and ‘consider’ for weak recommendations).
- The involvement of patients (and their carers if needed) in decisions on treatment and care.
- Consistency with NICE's standard advice on recommendations about drugs, waiting times and ineffective interventions (see Section 9.3 in The guidelines manual (2012)125).
The main considerations specific to each recommendation are outlined in the ‘Recommendations and link to evidence’ sections within each chapter.
3.5.1. Research recommendations
When areas were identified for which good evidence was lacking, the GDG considered making recommendations for future research. Decisions about inclusion were based on factors such as:
- the importance to patients or the population
- national priorities
- potential impact on the NHS and future NICE guidance
- ethical and technical feasibility.
3.5.2. Validation process
This guidance is subject to a 6-week public consultation and feedback as part of the quality assurance and peer review of the document. All comments received from registered stakeholders are responded to in turn and posted on the NICE website.
3.5.3. Updating the guideline
A formal review of the need to update a guideline is usually undertaken by NICE after its publication. NICE will conduct a review to determine whether the evidence base has progressed significantly to alter the guideline recommendations and warrant an update.
3.5.4. Disclaimer
Healthcare providers need to use clinical judgement, knowledge and expertise when deciding whether it is appropriate to apply guidelines. The recommendations cited here are a guide and may not be appropriate for use in all situations. The decision to adopt any of the recommendations cited here must be made by practitioners in light of individual patient circumstances, the wishes of the patient, clinical expertise and resources.
The National Clinical Guideline Centre disclaims any responsibility for damages arising out of the use or non-use of this guideline and the literature used in support of this guideline.
3.5.5. Funding
The National Clinical Guideline Centre was commissioned by the National Institute for Health and Care Excellence to undertake the work on this guideline.
Publication Details
Copyright
Publisher
National Institute for Health and Care Excellence (NICE), London
NLM Citation
National Clinical Guideline Centre (UK). Drug Allergy: Diagnosis and Management of Drug Allergy in Adults, Children and Young People. London: National Institute for Health and Care Excellence (NICE); 2014 Sep. (NICE Clinical Guidelines, No. 183.) 3, Methods.

