

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Guideline Centre (UK). Multimorbidity: Assessment, Prioritisation and Management of Care for People with Commonly Occurring Multimorbidity. London: National Institute for Health and Care Excellence (NICE); 2016 Sep. (NICE Guideline, No. 56.)

Multimorbidity: Assessment, Prioritisation and Management of Care for People with Commonly Occurring Multimorbidity.
Show detailsThis chapter sets out in detail the methods used to review the evidence and to develop the recommendations that are presented in subsequent chapters of this guideline. This guidance was developed in accordance with the methods outlined in the NICE guidelines manual, 2012 and 2014 versions.169,173
Sections 4.1 to 4.2 describe the process used to identify and review clinical evidence (summarised in Figure 1), Sections 4.1 and 4.3.6 describe the process used to identify and review the health economic evidence, and Section 4.5 describes the process used to develop recommendations.
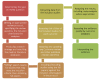
Figure 1
Step-by-step process of review of evidence in the guideline.
4.1. Developing the review questions and outcomes
Review questions were developed using a PICO framework (patient, intervention, comparison and outcome) for intervention reviews; using a framework of population, index tests, reference standard and target condition for reviews of diagnostic test accuracy; and using population, presence or absence of factors under investigation (for example prognostic factors) and outcomes for prognostic reviews.
This use of a framework guided the literature searching process, critical appraisal and synthesis of evidence, and facilitated the development of recommendations by the GDG. The review questions were drafted by the NCGC technical team and refined and validated by the GDG. The questions were based on the key clinical areas identified in the scope (Appendix A).
A total of 18 review questions were identified.
Full literature searches, critical appraisals and evidence reviews were completed for all the specified review questions.
Table 1Review questions
| Chapter | Type of review | Review questions | Outcomes |
|---|---|---|---|
| Qualitative | What principles are important for assessing, prioritising and managing care for people with multimorbidity? | Themes as identified by the evidence | |
| Qualitative | What are barriers to healthcare professionals optimising care for people with multimorbidity? | Themes as identified by the evidence | |
| Prognostic risk factor | What risk tool best identifies people with multimorbidity who are at risk of unplanned hospital admissions? | Unplanned hospital admissions (max time point=3 years) Statistical outputs may include:
| |
| Prognostic risk tool | What risk tool best identifies people with multimorbidity who are at risk of reduced health-related quality of life? | Reductions in health related quality of life (max time point=3 years) Statistical outputs may include:
| |
| Prognostic risk tool | What risk tool best identifies people with multimorbidity who are at risk of admission to care facility? | Admission to care facility (max time point = 3 years) Statistical outputs may include:
| |
| Prognostic risk tool | What risk tool best identifies people with multimorbidity who are at risk of reduced life expectancy? | Mortality Statistical outputs may include:
| |
| Prognostic risk factor | Is polypharmacy associated with a greater risk of unplanned hospital admissions amongst people with multimorbidity? | Unplanned hospital admissions at ≥ 1 year Statistical outputs may include:
| |
| Prognostic risk factor | Is polypharmacy associated with a greater risk of reductions in health-related quality of life amongst people with multimorbidity? | Health-related quality of life at ≥ 1 year Statistical outputs may include: Sensitivity, specificity, C-statistic, R2, beta coefficients, OR/RR, HR, MD will be extracted if no sensitivity/specificity data | |
| Prognostic risk factor | Is polypharmacy associated with a greater risk of admission to care facility amongst people with multimorbidity? | Admission to care facility at ≥ 1 year. Statistical outputs may include: Sensitivity, specificity, C-statistic, R2, beta coefficients, OR/RR, HR, MD will be extracted if no sensitivity/specificity data | |
| Prognostic risk factor | Is polypharmacy associated with a greater risk of mortality amongst people with multimorbidity? | Mortality at ≥ 1 year. Statistical outputs may include: Sensitivity, specificity, C-statistic, R2, beta coefficients, OR/RR, HR, MD will be extracted if no sensitivity/specificity data | |
| Diagnostic test accuracy | What is the most accurate tool for assessing frailty? | Sensitivity, specificity, C-statistic | |
| Questionnaire performance | How can treatment burden be assessed? | Reliability Validity Reproducibility Responsiveness Interpretability Time to complete User friendliness | |
| Bespoke review | How might data from condition-specific guidance best be used and presented to inform a ranking of treatments based on absolute risk and benefit and time to achieve benefits? |
| |
| Intervention | What is the clinical and cost-effectiveness of stopping antihypertensive treatment? | Critical:
| |
| Intervention | What is the clinical and cost effectiveness of stopping drugs for osteoporosis? | Critical:
| |
| Intervention | What is the clinical and cost effectiveness of stopping statin treatment? | Critical:
Myalgia (dichotomous) | |
| Intervention | What models of care improve outcomes in patients with multimorbidity? | Critical:
| |
| Intervention | What is the clinical and cost-effectiveness of self-management and expert patient programmes for people with multimorbidity? | Critical: Health-related quality of life (continuous) Mortality (time to event/dichotomous) Functional outcomes (mobility, activities of daily living) (continuous) Patient and carer satisfaction (continuous) Unplanned hospital admissions (dichotomous) Length of hospital stay (continuous) Important Continuity metrics (continuous) Patient/carer treatment burden (continuous) Patient self-efficacy (continuous) | |
| Intervention | What format of encounters with healthcare professionals improves outcomes for people with multimorbidity? | Critical
|
4.2. Searching for evidence
4.2.1. Clinical literature search
Systematic literature searches were undertaken to identify the published clinical evidence relevant to the review questions. Searches were undertaken according to the parameters stipulated within the NICE guidelines manual.169 Databases were searched using relevant medical subject headings, free-text terms and study-type filters where appropriate. Where possible, searches were restricted to articles published in English. Studies published in languages other than English were not reviewed. All searches were conducted in Medline, Embase, and The Cochrane Library. Additional subject specific databases were used for some questions: AMED for models of care; CINAHL for barriers, models or care and burden of treatment; PsycINFO for barriers and burden of treatment. All searches were updated on 4 January 2016. One additional paper 44 published after this date was included following stakeholder consultation and this is discussed in section 7.4.
Search strategies were quality assured by cross-checking reference lists of highly relevant papers, analysing search strategies in other systematic reviews, and asking GDG members to highlight any additional studies. Searches were quality assured by a second information scientist before being run. The questions, the study types applied, the databases searched and the years covered can be found in Appendix G.
The titles and abstracts of records retrieved by the searches were sifted for relevance, with potentially significant publications obtained in full text. These were assessed against the inclusion criteria. Reference lists for papers that met the inclusion criteria were checked for further potentially relevant papers. These papers were obtained in full text and assessed against the inclusion criteria.
During the scoping stage, a search was conducted for guidelines and reports on the websites listed below from organisations relevant to the topic.
- Guidelines International Network database (www.g-i-n.net)
- National Guideline Clearing House (www.guideline.gov)
- National Institute for Health and Care Excellence (NICE) (www.nice.org.uk)
- National Institutes of Health Consensus Development Program (consensus.nih.gov)
- NHS Evidence Search (www.evidence.nhs.uk).
All references sent by stakeholders were considered. Searching for unpublished literature was not undertaken. The NCGC and NICE do not have access to drug manufacturers' unpublished clinical trial results, so the clinical evidence considered by the GDG for pharmaceutical interventions may be different from that considered by the MHRA and European Medicines Agency for the purposes of licensing and safety regulation.
4.2.2. Health economic literature search
Systematic literature searches were also undertaken to identify health economic evidence within published literature relevant to the review questions. The evidence was identified by conducting a broad search relating to multimorbidity in the: NHS Economic Evaluation Database (NHS EED), the Health Technology Assessment database (HTA) and the Health Economic Evaluations Database (HEED) with no date restrictions (NHS EED ceased to be updated after March 2015; HEED was used for searches up to December 2014 but subsequently ceased to be available). Additionally, the search was run on Medline and Embase using a health economic filter, from 2013, to ensure recent publications that had not yet been indexed by the economic databases were identified. This was supplemented by additional searches that looked for economic papers specifically relating to models of care, holistic assessment, burden of treatment and stopping treatments on Medline, Embase, NHS EED, HTA and HEED as it became apparent that some papers in this area had not been identified by the first search. Where possible, searches were restricted to articles published in English. Studies published in languages other than English were not reviewed.
4.3. Identifying and analysing evidence of effectiveness
Research fellows conducted the tasks listed below, which are described in further detail in the rest of this section:
- Identified potentially relevant studies for each review question from the relevant search results by reviewing titles and abstracts. Full papers were then obtained.
- Reviewed full papers against pre-specified inclusion and exclusion criteria to identify studies that addressed the review question in the appropriate population, and reported on outcomes of interest (review protocols are included in Appendix C).
- Critically appraised relevant studies using appropriate study design checklist as specified in the NICE guidelines manual169,173. Prognostic risk factor reviews were appraised using QUIPS104,105 prognostic risk tool reviews were appraised using PROBAST, qualitative studies were critically appraised using NCGC checklists, and previously published guidelines were appraised using AGREE II.
- Extracted key information about interventional study methods and results using ‘Evibase’, NCGC's purpose-built software. Evibase produces summary evidence tables, including critical appraisal ratings. Key information about non-interventional study methods and results was manually extracted onto standard evidence tables and critically appraised separately (evidence tables are included in Appendix H).
- Generated summaries of the evidence by outcome. Outcome data were combined, analysed and reported according to study design:
- Randomised data for intervention reviews were meta-analysed where appropriate and reported in GRADE profiles. Where meta-analysis was not appropriate due to heterogeneity across studies, data from individual studies was presented separately.
- Diagnostic accuracy and prognostic data were meta-analysed where appropriate and reported in adapted GRADE profile tables. Where meta-analysis was not appropriate due to heterogeneity across studies, data from individual studies was presented separately.
- Qualitative data was summarised across studies where appropriate and reported in themes.
- Questionnaire performance data was presented as a range of values in adapted GRADE profiles.
- A sample of a minimum of 10% of the abstract lists of the first 3 sifts by new reviewers and those for complex review questions (for example, prognostic reviews) were double-sifted by a senior research fellow and any discrepancies were rectified. All of the evidence reviews were quality assured by a senior research fellow. This included checking:
- papers were included or excluded appropriately
- a sample of the data extractions
- correct methods were used to synthesise data
- a sample of the risk of bias assessments.
4.3.1. Inclusion and exclusion criteria
The inclusion and exclusion of studies was based on the criteria defined in the review protocols, which can be found in Appendix C. Excluded studies by review question (with the reasons for their exclusion) are listed in Appendix L The GDG was consulted about any uncertainty regarding inclusion or exclusion.
The key population inclusion criterion, relevant across the majority of the reviews in the guideline, was adults with multimorbidity. Multimorbidity was defined as the presence of two or more chronic conditions where these included at least one physical health condition. The key population exclusion criterion was people without multimorbidity, or people with multimorbidity with two or more mental health conditions without a coexisting physical health condition.
During development, it was noted that the majority of papers identified in literature searches did not specify whether the study population was multimorbid, or reported baseline characteristics that were unclear or unreliable measures of multimorbidity. The GDG agreed a standard for including papers without clear reporting of the multimorbidity of the population in a review, and under what circumstances these would be downgraded for indirectness as part of the quality process. This standard was intended to maximise the likelihood that papers included in the reviews were including people with multimorbidity, while also not excluding the vast majority of evidence that was identified. The standard used across the majority of the reviews is as follows:
Where papers clearly reported the proportion of people in the study sample who were multimorbid
- a paper was included if >95% of the population were multimorbid
- a paper was included if 80%-95% of the population were multimorbid and was downgraded once for indirectness.
- A paper was excluded if <80% of the population were multimorbid
Where papers did not clearly report the proportion of people in the study sample who were multimorbid
- A paper was included if the study sample was an older adult population (>65 years) and downgraded for indirectness. This standard is based on evidence that approximately 70% of older adults have two or more comorbidities. Papers were excluded if other baseline characteristics indicated that the population was not multimorbid.
- A paper may be included if the reviewer believed that the population is likely to be multimorbid based on the study characteristics reported in the paper. This included consideration of the population characteristics (for example, proportion of study population identified as frail; place of residence) and the study characteristics (for example, study aims and settings). These decisions were agreed with the GDG.
The GDG discussed reliable metrics of multimorbidity. The GDG agreed that the following metrics were not reliable indices of multimorbidity and papers could not be included based on these measures; (i) disease counts (for example, the Charlson comorbidity index) (ii) the mean number of conditions in the study sample, (iii) the N and % of participants with each single condition. These metrics were identified as being unreliable as they do not account for the propensity for conditions to ‘cluster’; such that individuals with one long-term condition are more likely than the general population to develop further long-term conditions.
In some cases, the standard was adjusted according to the need of the review. For example, studies with older adults where the proportion of the study sample with multimorbidity was unclear were not downgraded for indirectness if the GDG felt that this would not contribute to a difference in the effect size. Any alterations to the standard, and the rationale for this, is explained in the introduction for each of the reviews. Further information on the way papers were assessed for indirectness is explained later in this chapter (section 4.3.4).
Literature reviews, abstracts, posters, letters, editorials, comment articles, unpublished studies and studies not in English were excluded.
4.3.2. Type of studies
Randomised trials, observational studies (including diagnostic, prognostic, and questionnaire performance studies), qualitative studies, and previously published guidelines were included in the evidence reviews as appropriate.
For all intervention reviews in this guideline, parallel randomised controlled trials (RCTs) were prioritised for inclusion because they are considered the most robust type of study design that can produce an unbiased estimate of the intervention effects. For each intervention review, the GDG considered whether non-randomised trials were appropriate for inclusion. In all instances the GDG felt that RCTs would provide a better standard of evidence and therefore decided to only include non-randomised trials if no RCTs were included. No non-randomised trials were included in the guideline.
For diagnostic review questions, prospective and retrospective cohort studies in which the index test(s) and the reference standard test are applied to the same patients in a cross-sectional design were included. For prognostic review questions, prospective and retrospective cohort studies were included. Case–control studies were not included.
Two types of qualitative review were used in this guideline.
- One of these reviews sought the perspectives of individuals with multimorbidity, their carers, and healthcare professionals who provide care for people with multimorbidity. This review included interview and focus group studies.
- A separate review sought to identify principles for the care of people with multimorbidity that are recommended by experts in the care of multimorbidity, including people with multimorbidity, their carers, and healthcare professionals who care for people with multimorbidity. This review examined included reported advice and recommendations from already published guidelines relevant to the care of people with multimorbidity, including NICE guidelines, guidelines published by other recognised professional health groups, and other publications where the primary aim was to report recommendations for clinical practice.
In this guideline one questionnaire performance review was conducted to evaluate the performance of questionnaires where there was no established reference standard (gold standard) with which to derive diagnostic accuracy data. Cross-sectional, retrospective and prospective cohort studies were included.
Please refer to the review protocols in Appendix C for full details on the study design of studies selected for each review question.
4.3.3. Methods of combining clinical studies
4.3.3.1. Data synthesis for intervention reviews
Where possible, meta-analyses were conducted using Cochrane Review Manager (RevMan5)1 software to combine the data given in all studies for each of the outcomes of interest for the review question.
For some questions, the GDG specified that data should be stratified, meaning that studies that varied on a particular factor were not combined and analysed together. Where stratification was used, this is documented in the individual question protocols (see Appendix C). If additional strata were used this led to sub-strata (for example, 2 stratification criteria would lead to 4 sub-strata categories, or 3 stratification criteria would lead to 9 sub-strata categories) which would be analysed separately.
4.3.3.1.1. Analysis of different types of data
Dichotomous outcomes
Fixed-effects (Mantel-Haenszel) techniques (using an inverse variance method for pooling) were used to calculate risk ratios (relative risk, RR) for the binary outcomes, which included:
- mortality
- adverse events
- resource use.
The absolute risk difference was also calculated using GRADEpro98 software, using the median event rate in the control arm of the pooled results.
For binary variables where there were zero events in either arm or a less than 1% event rate, Peto odds ratios, rather than risk ratios, were calculated. Peto odds ratios are more appropriate for data with a low number of events.
Where sufficient information was provided, hazard ratios were calculated in preference for outcomes such as mortality where the time to the event occurring was important for decision-making. Where incomplete data was reported in a paper to extract Hazard Ratios, these were calculated according to established methods.228 Hazard ratio data was pooled using the generic inverse variance method in Cochran Review Manager (RevMan51 software).
Continuous outcomes
Continuous outcomes were analysed using an inverse variance method for pooling weighted mean differences. These outcomes included:
- heath-related quality of life (HRQoL)
- length of stay in hospital
- symptom scales (such as visual analogue scale)
- function and activities of daily living.
Where the studies within a single meta-analysis had different scales of measurement, standardised mean differences were used (providing all studies reported either change from baseline or final values rather than a mixture of both); each different measure in each study was ‘normalised’ to the standard deviation value pooled between the intervention and comparator groups in that same study.
The means and standard deviations of continuous outcomes are required for meta-analysis. However, in cases where standard deviations were not reported, the standard error was calculated if the p values or 95% confidence intervals (95% CI) were reported, and meta-analysis was undertaken with the mean and standard error using the generic inverse variance method in Cochrane Review Manager (RevMan51 software). Where p values were reported as ‘less than’, a conservative approach was undertaken. For example, if a p value was reported as ‘p≤0.001’, the calculations for standard deviations were based on a p value of 0.001. If these statistical measures were not available then the methods described in Section 16.1.3 of the Cochrane Handbook (version 5.1.0, updated March 2011) were applied.
4.3.3.1.2. Generic inverse variance
If a study reported only the summary statistic and 95% CI the generic-inverse variance method was used to enter data into RevMan5.1 If the control event rate was reported this was used to generate the absolute risk difference in GRADEpro.98 If multivariate analysis was used to derive the summary statistic but no adjusted control event rate was reported no absolute risk difference was calculated.
4.3.3.1.3. Heterogeneity
Statistical heterogeneity was assessed for each meta-analysis estimate by considering the chi-squared test for significance at p<0.1 or an I-squared (I2) inconsistency statistic (with an I-squared value of more than 50% indicating significant heterogeneity) as well as the distribution of effects. Where significant heterogeneity was present, predefined subgrouping of studies was carried out according to subgroup categories specified a priori on the protocol by the GDG (see Appendix C).
If the subgroup analysis resolved heterogeneity within all of the derived subgroups, then each of the derived subgroups were adopted as separate outcomes (providing at least 1 study remained in each subgroup. Assessments of potential differences in effect between subgroups were based on the chi-squared tests for heterogeneity statistics between subgroups. Any subgroup differences were interpreted with caution as separating the groups breaks the study randomisation and as such is subject to uncontrolled confounding.
If all predefined strategies of subgrouping were unable to explain statistical heterogeneity within each derived subgroup, then a random effects (DerSimonian and Laird) model was employed to the entire group of studies in the meta-analysis. A random-effects model assumes a distribution of populations, rather than a single population. This leads to a widening of the confidence interval around the overall estimate, thus providing a more realistic interpretation of the true distribution of effects across more than 1 population. If, however, the GDG considered the heterogeneity was so large that meta-analysis was inappropriate, then the results were described narratively.
4.3.3.2. Data synthesis for diagnostic test accuracy reviews
For diagnostic test accuracy studies, a positive result on the index test was found if the patient had values of the measured quantity above or below a threshold value, and different thresholds could be used. The thresholds were pre-specified by the GDG including whether or not data could be pooled across a range of thresholds. Diagnostic test accuracy measures used in the analysis were: area under the receiver operating characteristics (ROC) curve (AUC or C-statistic), and, for different thresholds (if appropriate), sensitivity and specificity. The threshold of a diagnostic test is defined as the value at which the test can best differentiate between those with and without the target condition. In practice this varies amongst studies. If a test has a high sensitivity then very few people with the condition will be missed (few false negatives). For example, a test with a sensitivity of 97% will only miss 3% of people with the condition. Conversely, if a test has a high specificity then few people without the condition would be incorrectly diagnosed (few false positives). For example, a test with a specificity of 97% will only incorrectly diagnose 3% of people who do not have the condition as positive. For each review, the GDG discussed the relative importance of sensitivity versus specificity, taking into consideration the clinical context of the review. Coupled forest plots of sensitivity and specificity with their 95% CIs across studies (at various thresholds) were produced for each test, using RevMan5.1 In order to do this, 2×2 tables (the number of true positives, false positives, true negatives and false negatives) were directly taken from the study if given, or else were derived from raw data or calculated from the set of test accuracy statistics.
Diagnostic meta-analysis was considered but was not conducted due to insufficient data. Evidence was presented individually, or as the median sensitivity and specificity where more than one study reported evidence for the same tool. If an even number of studies were reported the results of the study with the lower specificity value of the 2 middle studies was reported, alongside the full range of CIs from all studies.
Heterogeneity or inconsistency amongst studies was visually inspected in the forest plots.
Area under the ROC curve (AUC) data for each study were also plotted on a graph, for each diagnostic test. The AUC describes the overall diagnostic accuracy across the full range of thresholds. The following criteria were used for evaluating AUCs:
- ≤0.50: worse than chance
- 0.50–0.60: very poor
- 0.61–0.70: poor
- 0.71–0.80: moderate
- 0.81–0.92: good
- 0.91–1.00: excellent or perfect test.
Heterogeneity or inconsistency amongst studies was visually inspected.
4.3.3.3. Data synthesis for prognostic factor reviews
Evidence on the risk prediction of risk factors (discrimination data) were prioritised for inclusion, as these data can indicate the impact of using a risk factor in clinical practice to identify people who may be at risk of the outcome (that is, the sensitivity and specificity of the tool, as explained above (section 4.3.3.2). In addition, odds ratios (ORs), risk ratios (RRs) or hazard ratios (HRs), with their 95% confidence intervals (95% CIs) for the effect of the pre-specified prognostic factors were extracted from the studies. These data indicate the strength of the association between the risk factor and the outcome (for example, people with a threshold of x and above of a risk factors have twice the risk of the outcome than people under the x threshold of the risk factor). This data only provides an indication of the overall trend in relationship between the risk factor and outcome, and does not account for the fact that this relationship can vary between individuals and across populations and settings. Studies were only pooled if the GDG believed that the population, setting, and outcome were sufficiently similar across studies. Studies of lower risk of bias were preferred, taking into account the analysis and the study design. In particular, prospective cohort studies with a pre-specified threshold of the risk factor were preferred.
4.3.3.1. Data synthesis for risk prediction tools
For evidence reviews on risk prediction tools, results were presented separately for discrimination and calibration. The discrimination data were analysed according to the principles outlined under the section on data synthesis for diagnostic accuracy studies. As explained above (data synthesis for prognostic factor reviews), discrimination data can indicate the clinical impact of using a risk prediction tool in clinical practice, and therefore these data were prioritised for inclusion and decision-making. Calibration data for example, R2, if reported was presented separately to the discrimination data. Meta-analysis was considered but not performed due to insufficient data reported for each of the risk prediction tools. The results were presented for each study separately along with the quality rating for the study. Inconsistency and imprecision were assessed consistent with methods used for diagnostic accuracy reviews.
4.3.3.2. Data synthesis for qualitative study reviews
For each included paper subthemes were identified and linked to a generic theme. An example of a subtheme identified in one review is ‘viewing the patient individualistically and holistically’ and this was linked to a broader generic theme of ‘Relationship between patients and healthcare professionals’. In some cases, subthemes related to more than 1 generic theme. A summary evidence table of generic themes and underpinning subthemes was produced, along with a narrative description of the evidence, and a summary of the quality of the evidence.
4.3.3.3. Data synthesis for questionnaire performance reviews
Results for questionnaires included in the questionnaire performance review were presented individually. These reviews are useful to evaluate the performance of questionnaires or other tools where there is no available reference (gold) standard for evaluating the principle outcome. Without diagnostic test accuracy data, it is necessary to evaluate the performance of questionnaires across a number of performance metrics; including reliability, validity, and metrics related to the utility and interpretation of the questionnaire in clinical practice. Guidance from the literature was used to inform the interpretation of performance data, which is summarised below:
Table 2Interpretation of performance data
| Performance metric | Threshold for good performance and/or guidance for interpretation |
|---|---|
| Internal reliability | Cronbach's alpha for the scale is between 0.70 and 0.95 |
| Construct validity | The authors make clear, a priori hypotheses (including direction) between the scale and more than one related measure; appropriate measures are assessed appropriately and acceptable analysis used; at least 75% of the results are consistent with these hypotheses |
| Reproducibility | A clear time period to assess test-retest reliability is used; the intraclass coefficient (ICC), weighted kappa or Pearson's correlation coefficient is greater than 0.70; adequate agreement between the repeated tests (as assessed by whether the smallest detectable change or limits of agreement is smaller than the minimally important change |
| Responsiveness | If the responsiveness ratio is at least 1.96 or the AUC at least 0.70 |
| Interpretability | The authors provide mean scores and standard deviations for relevant subgroups in the sample; the authors provide information on what change in score would be clinically meaningful (MIC) |
| Time to complete | The time to complete the questionnaire (mean, SD and range) is appropriate to the intended setting of use of the questionnaire |
| User friendliness | If quantitative data used to assess user friendliness, scores (mean, SD and range) on a validated questionnaire indicate questionnaire is acceptable to an appropriate number of people relevant to the target population (as decided by the GDG). If qualitative data used to assess user friendliness, themes identified demonstrate no significant concerns of using the questionnaire in the intended population (as decided by the GDG), |
4.3.4. Appraising the quality of evidence by outcomes
4.3.4.1. Intervention reviews
The evidence for outcomes from the included RCTs and were evaluated and presented using an adaptation of the ‘Grading of Recommendations Assessment, Development and Evaluation (GRADE) toolbox’ developed by the international GRADE working group (http://www.gradeworkinggroup.org/). The software (GRADEpro98) developed by the GRADE working group was used to assess the quality of each outcome, taking into account individual study quality and the meta-analysis results.
Each outcome was first examined for each of the quality elements listed and defined in Table 3.
Table 3
Description of quality elements in GRADE for intervention studies.
Details of how the 4 main quality elements (risk of bias, indirectness, inconsistency and imprecision) were appraised for each outcome are given below. Publication or other bias was only taken into consideration in the quality assessment if it was apparent.
4.3.4.1.1. Risk of bias
The main domains of bias for RCTs are listed in Table 4. Each outcome had its risk of bias assessed within each study first. For each study, if there were no risks of bias in any domain, the risk of bias was given a rating of 0. If there was risk of bias in just 1 domain, the risk of bias was given a ‘serious’ rating of −1, but if there was risk of bias in 2 or more domains the risk of bias was given a ‘very serious’ rating of −2. A weighted average score was then calculated across all studies contributing to the outcome, by taking into account the weighting of studies according to study precision. For example if the most precise studies tended to each have a score of −1 for that outcome, the overall score for that outcome would tend towards −1.
Table 4
Principle domains of bias in randomised controlled trials.
4.3.4.1.2. Indirectness
Indirectness refers to the extent to which the populations, interventions, comparisons and outcome measures are dissimilar to those defined in the inclusion criteria for the reviews. Indirectness is important when these differences are expected to contribute to a difference in effect size, or may affect the balance of harms and benefits considered for an intervention. As for the risk of bias, each outcome had its indirectness assessed within each study first. For each study, if there were no sources of indirectness, indirectness was given a rating of 0. If there was indirectness in just 1 source (for example in terms of population), indirectness was given a ‘serious’ rating of −1, but if there was indirectness in 2 or more sources (for example, in terms of population and treatment) the indirectness was given a ‘very serious’ rating of −2. A weighted average score was then calculated across all studies contributing to the outcome by taking into account study precision. For example, if the most precise studies tended to have an indirectness score of −1 each for that outcome, the overall score for that outcome would tend towards −1.
4.3.4.1.3. Inconsistency
Inconsistency refers to an unexplained heterogeneity of results for an outcome across different studies. When estimates of the treatment effect across studies differ widely, this suggests true differences in the underlying treatment effect, which may be due to differences in populations, settings or doses. When heterogeneity existed within an outcome (chi-squared p<0.1, or I2>50%), but no plausible explanation could be found, the quality of evidence for that outcome was downgraded. Inconsistency for that outcome was given a ‘serious’ score of −1 if the I2 was 50–74% and a ‘very serious’ score of −2 if the I2 was 75% or more.
If inconsistency could be explained based on pre-specified subgroup analysis (that is, each subgroup had an I2<50%), the GDG took this into account and considered whether to make separate recommendations on new outcomes based on the subgroups defined by the assumed explanatory factors. In such a situation the quality of evidence was not downgraded for those emergent outcomes.
Since the inconsistency score was based on the meta-analysis results, the score represented the whole outcome and so weighted averaging across studies was not necessary.
4.3.4.1.4. Imprecision
The criteria applied for imprecision were based on the 95% CIs for the pooled estimate of effect, and the minimal important differences (MID) for the outcome. The MIDs are the threshold for appreciable benefits and harms, separated by a zone either side of the line of no effect where there is assumed to be no clinically important effect. If either end of the 95% CI of the overall estimate of effect crossed 1 of the MID lines, imprecision was regarded as serious and a ‘serious’ score of −1 was given. This was because the overall result, as represented by the span of the confidence interval, was consistent with 2 interpretations as defined by the MID (for example, both no clinically important effect and clinical benefit were possible interpretations). If both MID lines were crossed by either or both ends of the 95% CI then imprecision was regarded as very serious and a ‘very serious’ score of −2 was given. This was because the overall result was consistent with all 3 interpretations defined by the MID (no clinically important effect, clinical benefit and clinical harm). This is illustrated in Figure 2. As for inconsistency, since the imprecision score was based on the meta-analysis results, the score represented the whole outcome and so weighted averaging across studies was not necessary.
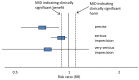
Figure 2
Illustration of precise and imprecise outcomes based on the 95% CI of dichotomous outcomes in a forest plot (Note that all 3 results would be pooled estimates, and would not, in practice, be placed on the same forest plot).
The position of the MID lines is ideally determined by values reported in the literature. ‘Anchor-based’ methods aim to establish clinically meaningful changes in a continuous outcome variable by relating or ‘anchoring’ them to patient-centred measures of clinical effectiveness that could be regarded as gold standards with a high level of face validity. For example, a MID for an outcome could be defined by the minimum amount of change in that outcome necessary to make patients feel their quality of life had ‘significantly improved’. MIDs in the literature may also be based on expert clinician or consensus opinion concerning the minimum amount of change in a variable deemed to affect quality of life or health. For binary variables, any MIDs reported in the literature will inevitably be based on expert consensus, as such MIDs relate to all-or-nothing population effects rather than measurable effects on an individual, and so are not amenable to patient-centred ‘anchor’ methods.
In this guideline, MIDs found in the literature were used to assess imprecision for the EQ-5D and SF-36 measures of health-related quality of life. These values are displayed below:
Table 5MIDs used to assess imprecision for the EQ-5D and SF-36 measures
| MIDs for assessing between group differences Outcome | MID for imprecision | MID for clinical importance | Source |
|---|---|---|---|
| SF-36^ | Physical component summary: 2 Mental component summary: 3 Physical functioning: 3 Role-physical: 3 Bodily pain: 3 General health: 2 Vitality: 2 Social functioning: 3 Role-emotional: 4 Mental health: 3 | User's manual for the SF-36v2 Health Survey, Third Edition | |
| EQ5D* | GRADE defaults | 0.03 | NICE agreed for use in Low Back Pain & Low back Pain GDG opinion |
- ^
Note: the SF-12 manual does not specify MIDs. It does however signpost to the SF-36 manual for guidance on interpretation, therefore in this guideline we used the same MIDs for the SF-12.
- *
Note: this is not based on the literature and was a pragmatic decision for this guideline based on the SF-36 MIDs.
In the absence of values identified in the literature, the alternative approach to deciding on MID levels is the ‘default’ method, as follows:
- For categorical outcomes the MIDs were taken to be RRs of 0.75 and 1.25. For ‘positive’ outcomes such as ‘patient satisfaction’, the RR of 0.75 is taken as the line denoting the boundary between no clinically important effect and a clinically significant harm, whilst the RR of 1.25 is taken as the line denoting the boundary between no clinically important effect and a clinically significant benefit. For ‘negative’ outcomes such as ‘stroke’, the opposite occurs, so the RR of 0.75 is taken as the line denoting the boundary between no clinically important effect and a clinically significant benefit, whilst the RR of 1.25 is taken as the line denoting the boundary between no clinically important effect and a clinically significant harm.
- For mortality and admission to care home any change was considered to be clinically important and the imprecision was assessed on the basis of whether the confidence intervals crossed the line of no effect, that is, whether the result was consistent with both benefit and harm.
- For continuous outcome variables the MID was taken as half the median baseline standard deviation of that variable, across all studies in the meta-analysis. Hence the MID denoting the minimum clinically significant benefit was positive for a ‘positive’ outcome (for example, a quality of life measure where a higher score denotes better health), and negative for a ‘negative’ outcome (for example, a visual analogue scale [VAS] pain score). Clinically significant harms will be the converse of these. If baseline values are unavailable, then half the median comparator group standard deviation of that variable will be taken as the MID.
- If standardised mean differences have been used, then the MID will be set at the absolute value of +0.5. This follows because standardised mean differences are mean differences normalised to the pooled standard deviation of the 2 groups, and are thus effectively expressed in units of ‘numbers of standard deviations’. The 0.5 MID value in this context therefore indicates half a standard deviation, the same definition of MID as used for non-standardised mean differences.
The default MID value was subject to amendment after discussion with the GDG. If the GDG decided that the MID level should be altered, after consideration of absolute as well as relative effects, this was allowed, provided that any such decision was not influenced by any bias towards making stronger or weaker recommendations for specific outcomes.
4.3.4.1.5. Overall grading of the quality of clinical evidence
Once an outcome had been appraised for the main quality elements, as above, an overall quality grade was calculated for that outcome. The scores (0, −1 or −2) from each of the main quality elements were summed to give a score that could be anything from 0 (the best possible) to −8 (the worst possible). However scores were capped at −3. This final score was then applied to the starting grade that had originally been applied to the outcome by default, based on study design. All RCTs started as High and the overall quality became Moderate, Low or Very Low if the overall score was −1, −2 or −3 points respectively. The significance of these overall ratings is explained in Table 6. The reasons for downgrading in each case were specified in the footnotes of the GRADE tables.
Table 6
Overall quality of outcome evidence in GRADE.
Observational interventional studies started at Low, and so a score of −1 would be enough to take the grade to the lowest level of Very Low. Observational studies could, however, be upgraded if there were all of: a large magnitude of effect, a dose-response gradient, and if all plausible confounding would reduce the demonstrated effect.
4.3.4.2. Diagnostic studies
Risk of bias and indirectness of evidence for diagnostic data were evaluated by study using the Quality Assessment of Diagnostic Accuracy Studies version 2 (QUADAS-2) checklists (see Appendix H in the NICE guidelines manual 2014169). Risk of bias and applicability in primary diagnostic accuracy studies in QUADAS-2 consists of 4 domains (see Table 7):
Table 7
Summary of QUADAS-2 with list of signalling, risk of bias and applicability questions.
- patient selection
- index test
- reference standard
- flow and timing.
4.3.4.2.1. Inconsistency
Inconsistency refers to an unexplained heterogeneity of results for an outcome across different studies. Inconsistency was assessed by inspection of the specificity value (based on the primary measure) using the point estimates and 95% CIs of the individual studies on the forest plots. Particular attention was placed on values above or below 50% (diagnosis based on chance alone) and the threshold set by the GDG (the threshold above which it would be acceptable to recommend a test). For example, the GDG might have set a threshold of 90% as an acceptable level to recommend a test. The evidence was downgraded by 1 increment if the individual studies varied across 2 areas (for example, 50–90% and 90–100%) and by 2 increments if the individual studies varied across 3 areas (for example, 0–50%, 50–90% and 90–100%).
4.3.4.2.2. Imprecision
Diagnostic meta-analysis was not conducted in this guideline, and imprecision was assessed according to the range of point estimates or, if only one study contributed to the evidence, the 95% CI around the single study. As a general rule (after discussion with the GDG) a variation of 0–20% was considered precise, 20–40% serious imprecision, and >40% very serious imprecision. Imprecision was assessed on the primary outcome measure for decision-making.
4.3.4.2.3. Overall grading
Quality rating started at High for both prospective and retrospective studies, and each major limitation (risk of bias, indirectness, inconsistency and imprecision) brought the rating down by 1 increment to a minimum grade of Very Low, as explained for intervention reviews.
4.3.4.3. Prognostic risk factor studies
In this guideline, the quality of evidence for prognostic risk factor studies was evaluated according to an amended QUIPS checklist104,105 which is reported in Table 8. The QUIPS was amended to remove the section on the adequate control of confounding. This is because for the polypharmacy reviews in this guideline, the unadjusted data was preferred and control of plausible confounding was not necessary. If data were meta-analysed the quality for pooled studies was presented. If the data was not pooled then a quality rating was presented for each study.
Table 8
Description of quality elements for prognostic risk factor studies.
Indirectness
Indirectness was assessed as for intervention studies.
Inconsistency
Inconsistency was assessed as for intervention studies.
Imprecision
The criteria applied for imprecision were based on the confidence intervals for the pooled estimate of effect. If either of the 95% confidence intervals of the overall estimate of effect crossed the null line then imprecision was regarded as serious and a ‘serious’ score of -1 was given. This was because the overall result, as represented by the span of the confidence intervals, was consistent with two conflicting interpretations as defined by the line of no effect (for example, predictive of either low or high risk of the outcome).
Overall grading
Because prognostic reviews were not usually based on multiple outcomes per study, quality rating was assigned by study. However if there was more than one outcome involved in a study, then the quality rating of the evidence statements for each outcome was adjusted accordingly. For example, if one outcome was based on an invalidated measurement method, but another outcome in the same study wasn't, the latter outcome would be graded one grade higher than the other.
Quality rating started at high for prospective and retrospective studies, and each major limitation brought the rating down by one increment to a minimum grade of VERY LOW, as explained for interventional studies. For prognostic studies, prediction tool studies for prognosis are regarded as the gold standard because RCTs are usually inappropriate for these types of review for ethical or pragmatic reasons. Furthermore if the study is looking at more than one risk factor of interest then randomisation would be inappropriate as it can only be applied to one of the risk factors.
4.3.4.4. Prognostic risk tool studies
Risk of bias and indirectness (applicability) of evidence for prognostic risk tool data was evaluated using the Prediction Study Risk of Bias Assessment tool (PROBAST)2 checklist, which is summarised in Table 9. PROBAST is still under development and the version used in this guideline was acquired from the study author and adapted. One item concerning whether all predictors were available at the time the risk tool would be used in practice was excluded from the risk of bias assessment, and instead was incorporated into an assessment of indirectness. Where the information required to complete PROBAST domains was not reported in publications, this was taken into account for the risk of bias assessment. If the majority of information was available but one domain had limited information there was no obligate downgrade for risk of bias. If more than one domain had limited or no information to inform it's assessment, the study was downgraded once for risk of bias. If very limited or no information was provided for the majority of domains for the study, it was downgraded twice for risk of bias. Ratings were derived for the validation of risk tools; no ratings were provided for the original development phase of the tools.
Table 9
Summary of PROBAST.
Inconsistency
Inconsistency refers to an unexplained heterogeneity of results for an outcome across different studies. Inconsistency was assessed by inspection of the specificity value (based on the primary measure) using the point estimates and confidence intervals of the individual studies on the forest plots. Particular attention was placed on values above or below 50% (prognostic accuracy based on chance alone) and the threshold set by the GDG (the threshold above which would be acceptable to recommend a test) – for example, the GDG might set a threshold of 90% as an acceptable level to recommend a test. The evidence was downgraded by 1 increment if the individual studies varied across 2 areas (for example, 50–90% and 90–100%) and by 2 increments if the individual studies varied across 3 areas (for example, 0–50%, 50–90% and 90–100%).
Imprecision
The judgement of precision was based on visual inspection of the confidence region around the summary sensitivity and specificity point from the diagnostic meta-analysis, if a diagnostic meta-analysis was conducted. Where a diagnostic meta-analysis was not conducted imprecision was assessed according to the range of point estimates or, if only one study contributed to the evidence, the confidence interval around the single study. As a rule of thumb (after discussion with the GDG) a variation of 0–20% was considered precise, 20–40% serious imprecision, and >40% very serious imprecision. Imprecision was assessed on the primary measure for decision-making
Overall grading
Because prognostic reviews were not usually based on multiple tools or outcomes per study, quality rating was assigned by study. However if there was more than one tool or outcome involved in a study, then the quality rating of the evidence statements for each tool and for each outcome was adjusted accordingly. For example, if one outcome was based on an invalidated measurement method, but another outcome in the same study wasn't, the latter outcome would be graded one grade higher than the other.
Quality rating started at HIGH for prospective and retrospective cohort studies, and each major limitation (risk of bias, indirectness, inconsistency and imprecision) brought the rating down by one increment to a minimum grade of VERY LOW, as explained for interventional studies.
4.3.4.5. Qualitative reviews
As explained above in Section 4.3.2, two types of qualitative reviews were included in this guideline. For the review where interviews and focus groups studies were included, the checklist summarised in Table 10 below was used to appraise the quality for each sub-theme. The overall quality rating for each theme is reported in a summary table in the evidence report.
Table 10
Summary of factors used to assess quality in qualitative studies.
In the other qualitative review we included recommendations from already published guidelines and other publications where the primary aim was to report recommendations for clinical practice. The quality of this evidence was assessed using AGREE II criteria.37 The AGREE II tool is used to appraise the quality of guidelines, and is comprised of 6 individual domains and an overall quality rating, summarised in Table 11 below. Consistent with the AGREE II approach, a quality rating for each domain was reported for every guideline. No summary quality rating was produced for the themes identified in the analysis.
Table 11
Summary of domains used to assess quality of published guidelines.
4.3.4.6. Questionnaire performance reviews
Risk of bias of evidence for questionnaire performance data was evaluated using the Questionnaire Bias Assessment Tool (Q-BAST)78, which is summarised in Table 12 below. Q-BAST consists of six domains, with risk of bias for each domain rated as high, low or unclear. An unclear rating was only given if there was insufficient information provided in the report to make a judgement. An overall rating for each questionnaire was derived, which represented the overall risk of bias across all six domains:
Table 12
Summary of Q-BAST with list of signalling questions.
Indirectness
Indirectness was assessed as for intervention studies.
4.3.5. Assessing clinical importance
The GDG assessed the evidence by outcome in order to determine if there was, or potentially was, a clinically important benefit, a clinically important harm or no clinically important difference between interventions. To facilitate this, binary outcomes were converted into absolute risk differences (ARDs) using GRADEpro98 software: the median control group risk across studies was used to calculate the ARD and its 95% CI from the pooled risk ratio.
To interpret the clinical evidence for EQ-5D and SF-36 health related quality of life outcomes, the default MIDs (as described in 4.3.4.1.4) were used to identify if the difference between the intervention and comparison indicated a clinical benefit or harm. For other outcomes where MIDs from the literature were not available, the GDG discussed and agreed on whether the point estimate of absolute effect indicated a clinical benefit, harm, or no benefit or harm for each critical outcome. For the critical outcomes of mortality and admission to care home, the GDG agreed that any change would be clinically important; that is, any reduction represented a clinical benefit and any increase represented a clinical harm.
An evidence summary table was produced to compile the GDG's assessments of clinical importance per outcome, alongside the evidence quality and the uncertainty in the effect estimate (imprecision).
4.3.6. Clinical evidence statements
Clinical evidence statements are summary statements that are included in each review chapter, and which summarise the key features of the clinical effectiveness evidence presented. For reviews in this guideline with a limited amount of clinical effectiveness evidence, the evidence statements are presented by outcome and encompass the following key features of the evidence:
- The number of studies and the number of participants for a particular outcome.
- An indication of the direction of clinical importance (if one treatment is beneficial or harmful compared to the other or whether there is no difference between the 2 tested treatments).
- A description of the overall quality of the evidence (GRADE overall quality).
Some of the reviews in this guideline contained a large amount of clinical effectiveness evidence (for example, where a large number of different risk tools were evaluated). For these reviews, a summary of the clinical effectiveness evidence was provided, which encompassed the following key features of the evidence:
- The overall direction of the evidence (for example, the GDG's impression of the clinical effectiveness of the interventions identified and whether any interventions emerged as being strongly clinically beneficial or harmful across critical outcomes)
- Any variation in the direction or quality of the evidence (for example, if the evidence for an intervention was weaker or stronger in a particular strata or subgroup
- More detailed description of key evidence, such as that which was integral to the GDG's discussion and formulation of a recommendation (for example, interventions that emerged as strongly beneficial for people with multimorbidity); including the number of studies and participants for a particular outcome, and a description of the overall quality of the evidence (GRADE overall quality).
4.4. Identifying and analysing evidence of cost-effectiveness
The GDG is required to make decisions based on the best available evidence of both clinical effectiveness and cost-effectiveness. Guideline recommendations should be based on the expected costs of the different options in relation to their expected health benefits (that is, their ‘cost-effectiveness’) in addition to the total implementation cost.169
Health economic evidence was sought relating to the key clinical issues being addressed in the guideline. Health economists:
- Undertook a systematic review of the published economic literature.
- Undertook new cost-effectiveness analysis in priority areas.
4.4.1. Literature review
The health economists:
- Identified potentially relevant studies for each review question from the health economic search results by reviewing titles and abstracts. Full papers were then obtained.
- Reviewed full papers against pre-specified inclusion and exclusion criteria to identify relevant studies (see below for details).
- Extracted key information about the studies' methods and results into economic evidence tables (included in Appendix I).
- Generated summaries of the evidence in NICE economic evidence profile tables (included in the relevant chapter for each review question) – see below for details.
4.4.1.1. Inclusion and exclusion criteria
Full economic evaluations (studies comparing costs and health consequences of alternative courses of action: cost-utility, cost-effectiveness, cost-benefit and cost-consequences analyses) and comparative costing studies that addressed the review question in the relevant population were considered potentially includable as economic evidence.
Studies that only reported cost per hospital (not per patient), or only reported average cost-effectiveness without disaggregated costs and effects were excluded. Literature reviews, abstracts, posters, letters, editorials, comment articles, unpublished studies and studies not in English were excluded. Studies published before 1999 and studies from non-OECD countries or the USA were also excluded, on the basis that the applicability of such studies to the present UK NHS context is likely to be too low for them to be helpful for decision-making.
Remaining health economic studies were prioritised for inclusion based on their relative applicability to the development of this guideline and the study limitations. For example, if a high quality, directly applicable UK analysis was available, then other less relevant studies may not have been included. Where exclusions occurred on this basis, this is noted in the relevant section.
For more details about the assessment of applicability and methodological quality see Table 13 below and the economic evaluation checklist (Appendix G of the 2012 NICE guidelines manual173) and the health economics review protocol in Appendix D.
Table 13
Content of NICE economic evidence profile.
4.4.1.2. NICE economic evidence profiles
NICE economic evidence profile tables were used to summarise cost and cost-effectiveness estimates for the included health economic studies in each review chapter. The economic evidence profile shows an assessment of applicability and methodological quality for each economic study, with footnotes indicating the reasons for the assessment. These assessments were made by the health economist using the economic evaluation checklist from the NICE guidelines manual.173 It also shows the incremental costs, incremental effects (for example, quality-adjusted life years [QALYs]) and incremental cost-effectiveness ratio (ICER) for the base case analysis in the study, as well as information about the assessment of uncertainty in the analysis. See Table 13 for more details.
When a non-UK study was included in the profile, the results were converted into pounds sterling using the appropriate purchasing power parity.184
4.4.2. Undertaking new health economic analysis
As well as reviewing the published health economic literature for each review question, as described above, new health economic analysis was undertaken by the health economist in selected areas. Priority areas for new analysis were agreed by the GDG after formation of the review questions and consideration of the existing health economic evidence.
The GDG identified outpatient holistic assessment as the highest priority area for original health economic modelling. This area was prioritised as there was uncertainty around the cost effectiveness of holistic assessment as it increases costs but the evidence showed some benefits. More details on the original analysis are reported in Chapter 11 and Appendix N
The following general principles were adhered to in developing the cost-effectiveness analysis:
- The GDG was involved in the design of the model, selection of inputs and interpretation of the results.
- Model inputs were based on the systematic review of the clinical literature supplemented with other published data sources where possible.
- When published data were not available GDG expert opinion was used to populate the model.
- Model inputs and assumptions were reported fully and transparently.
- The results were subject to sensitivity analysis and limitations were discussed.
- The model was peer-reviewed by another health economist at the NCGC.
Full methods for the cost-effectiveness analysis for holistic assessment are described in Appendix N.
4.4.3. Cost-effectiveness criteria
NICE's report ‘Social value judgements: principles for the development of NICE guidance’ sets out the principles that GDGs should consider when judging whether an intervention offers good value for money.171 In general, an intervention was considered to be cost-effective (given that the estimate was considered plausible) if either of the following criteria applied:
- the intervention dominated other relevant strategies (that is, it was both less costly in terms of resource use and more clinically effective compared with all the other relevant alternative strategies), or
- the intervention cost less than £20,000 per QALY gained compared with the next best strategy.
If the GDG recommended an intervention that was estimated to cost more than £20,000 per QALY gained, or did not recommend one that was estimated to cost less than £20,000 per QALY gained, the reasons for this decision are discussed explicitly in the ‘Recommendations and link to evidence’ section of the relevant chapter, with reference to issues regarding the plausibility of the estimate or to the factors set out in ‘Social value judgements: principles for the development of NICE guidance’.171
When QALYs or life years gained are not used in the analysis, results are difficult to interpret unless one strategy dominates the others with respect to every relevant health outcome and cost.
4.4.4. In the absence of economic evidence
When no relevant published health economic studies were found, and a new analysis was not prioritised, the GDG made a qualitative judgement about cost-effectiveness by considering expected differences in resource use between options and relevant UK NHS unit costs, alongside the results of the review of clinical effectiveness evidence.
The UK NHS costs reported in the guideline are those that were presented to the GDG and were correct at the time recommendations were drafted. They may have changed subsequently before the time of publication. However, we have no reason to believe they have changed substantially.
4.5. Developing recommendations
Over the course of the guideline development process, the GDG was presented with:
- Evidence tables of the clinical and economic evidence reviewed from the literature. All evidence tables are in Appendices H and I.
- Summaries of clinical and economic evidence and quality (as presented in Chapters 5 - 12).
- Forest plots (Appendix K).
- A description of the methods and results of the cost-effectiveness analysis undertaken for the guideline (Appendix N).
Recommendations were drafted on the basis of the GDG's interpretation of the available evidence, taking into account the balance of benefits, harms and costs between different courses of action. This was either done formally in an economic model, or informally. Firstly, the net clinical benefit over harm (clinical effectiveness) was considered, focusing on the critical outcomes. When this was done informally, the GDG took into account the clinical benefits and harms when one intervention was compared with another. The assessment of net clinical benefit was moderated by the importance placed on the outcomes (the GDG's values and preferences), and the confidence the GDG had in the evidence (evidence quality). Secondly, the GDG assessed whether the net clinical benefit justified any differences in costs between the alternative interventions.
When clinical and economic evidence was of poor quality, conflicting or absent, the GDG drafted recommendations based on its expert opinion. The considerations for making consensus-based recommendations include the balance between potential harms and benefits, the economic costs compared to the economic benefits, current practices, recommendations made in other relevant guidelines, patient preferences and equality issues. The consensus recommendations were agreed through discussions in the GDG. The GDG also considered whether the uncertainty was sufficient to justify delaying making a recommendation to await further research, taking into account the potential harm of failing to make a clear recommendation (see Section 4.5.1 below).
The GDG considered the appropriate ‘strength’ of each recommendation. This takes into account the quality of the evidence but is conceptually different. Some recommendations are ’strong’ in that the GDG believes that the vast majority of healthcare and other professionals and patients would choose a particular intervention if they considered the evidence in the same way that the GDG has. This is generally the case if the benefits clearly outweigh the harms for most people and the intervention is likely to be cost-effective. However, there is often a closer balance between benefits and harms, and some patients would not choose an intervention whereas others would. This may happen, for example, if some patients are particularly averse to some side effect and others are not. In these circumstances the recommendation is generally weaker, although it may be possible to make stronger recommendations about specific groups of patients.
The GDG focused on the following factors in agreeing the wording of the recommendations:
- The actions health professionals need to take.
- The information readers need to know.
- The strength of the recommendation (for example the word ‘offer’ was used for strong recommendations and ‘consider’ for weaker recommendations).
- The involvement of patients (and their carers if needed) in decisions on treatment and care.
- Consistency with NICE's standard advice on recommendations about drugs, waiting times and ineffective interventions (see Section 9.2 in the 2014 NICE guidelines manual169).
The main considerations specific to each recommendation are outlined in the ‘Recommendations and link to evidence’ sections within each chapter.
4.5.1. Research recommendations
When areas were identified for which good evidence was lacking, the GDG considered making recommendations for future research. Decisions about the inclusion of a research recommendation were based on factors such as:
- the importance to patients or the population
- national priorities
- potential impact on the NHS and future NICE guidance
- ethical and technical feasibility.
4.5.2. Validation process
This guidance is subject to a 6-week public consultation and feedback as part of the quality assurance and peer review of the document. All comments received from registered stakeholders are responded to in turn and posted on the NICE website.
4.5.3. Updating the guideline
Following publication, and in accordance with the NICE guidelines manual, NICE will undertake a review of whether the evidence base has progressed significantly to alter the guideline recommendations and warrant an update.
4.5.4. Disclaimer
Healthcare providers need to use clinical judgement, knowledge and expertise when deciding whether it is appropriate to apply guidelines. The recommendations cited here are a guide and may not be appropriate for use in all situations. The decision to adopt any of the recommendations cited here must be made by practitioners in light of individual patient circumstances, the wishes of the patient, clinical expertise and resources.
The National Clinical Guideline Centre disclaims any responsibility for damages arising out of the use or non-use of this guideline and the literature used in support of this guideline.
4.5.5. Funding
The National Clinical Guideline Centre was commissioned by the National Institute for Health and Care Excellence to undertake the work on this guideline.
- Methods - Multimorbidity: Assessment, Prioritisation and Management of Care for ...Methods - Multimorbidity: Assessment, Prioritisation and Management of Care for People with Commonly Occurring Multimorbidity
Your browsing activity is empty.
Activity recording is turned off.
See more...