1.1. Disease Prevalence and Incidence
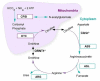
The urea cycle is responsible for the metabolism of nitrogen produced through the breakdown of protein and other nitrogen-containing molecules. It accomplishes this by converting ammonia to urea, which is excreted from the body ().1,2 Urea cycle disorders (UCDs) result from genetic mutations that cause defects in any of the five enzymes of the urea cycle in the liver: carbamoyl phosphate synthetase 1 (CPS1), ornithine transcarbamylase (OTC), argininosuccinate synthetase (ASS), argininosuccinate lyase, and arginase; in the co-factor producer N-acetyl glutamate synthetase; or in ornithine transporter and citrin. Deficiencies of CPS1, ASS, argininosuccinate lyase, arginase, N-acetyl glutamate synthetase, ornithine transporter, and citrin are inherited in an autosomal recessive manner, while OTC deficiencies are inherited in an X-linked manner.1,3 The incidence of UCDs is difficult to determine owing to the rarity of the condition and undiagnosed cases, but estimates ranging from one in 22,179 births to one in 53,717 births have been reported.4 The most recent estimate of incidence of UCDs for the US is around one in 35,000 births.5 Assuming the same incidence in Canada and using a birth rate of 380,863 live births per year, it is estimated that approximately 11 new cases of UCDs will be diagnosed each year in Canada.6 The incidence of OTC deficiency (one in 56,500 live births) is higher than other UCDs.5
The Urea Cycle. ARG = arginase; ASL = argininosuccinate lyase; ASS = argininosuccinate synthetase; ATP = adenosine triphosphate; CoA = coenzyme A; CPS1 = carbamoyl phosphate synthetase 1; ORNT1 = ornithine transporter; OTC = ornithine transcarbamylase. (more...)
Deficiencies in the urea cycle may result in excessive ammonia levels due to impaired metabolism, which can be life-threatening and result in permanent neurological damage if left untreated. Infants with a complete enzyme deficiency in a urea cycle (other than arginase) often present in the newborn period (neonatal-onset) with hyperammonemic coma, and the mortality rate is 50% after five years.7 Survivors often experience severe developmental delay and recurrent hyperammonemic episodes.8 Patients with partial deficiencies have variable clinical presentations and later onset, but still have a 10% risk of mortality and a significant risk of developmental disabilities.7,9 OTC deficiency affects males and females differently as a result of its X-linked inheritance, with affected males being more likely to present neonatally with severe hyperammonemia, and female carriers presenting with a later onset.5,10
UCDs are diagnosed using a combination of clinical parameters, laboratory parameters, family history, and genetic testing.8 Since hyperammonemia is the hallmark for most UCDs and may cause permanent damage, blood ammonia levels should be taken to evaluate a patient with a suspected UCD in an emergency setting if there is an unexplained change in consciousness, unusual neurological illness, liver failure, or suspected intoxication. If hyperammonemia is confirmed, plasma amino acids, blood or plasma acylcarnitines, urinary organic acids, and orotic acids should be determined, along with basic laboratory investigations, in order to differentiate between hyperammonemia due to inborn errors from other conditions. The specific UCD can be determined using laboratory parameters based on argininosuccinate, citrulline, arginine, ornithine, and orotic acid levels. For confirmation of diagnosis, genetic testing or enzymatic assays using liver biopsy samples should be performed.8 According to the Urea Cycle Disorders Consortium, an elevated plasma ammonia level of ≥ 150 μmol/L in neonates or > 100 μmol/L in older children and adults is a strong indication for the presence of a UCD.11
1.2. Standards of Therapy
Neurologic abnormalities and impaired cognitive function are significantly correlated with the frequency, severity, and duration of hyperammonemia; therefore, treatment should be initiated as soon as a diagnosis of a UCD is suspected and should proceed simultaneously with the diagnostic evaluation.10,12
Emergency management of patients in hyperammonemic coma resulting from UCD includes removing ammonia from the body using medications and/or dialysis, stopping protein intake and minimizing catabolism, and stimulating anabolism and uptake of nitrogen precursors by muscle.1,8,12 Medication for hyperammonemia consists of administering a combination of sodium phenylacetate and sodium benzoate (i.e., Ammonul used in Europe), arginine, citrulline (for OTC or CPS1 deficiency), and carglumic acid (for N-acetyl glutamate synthetase deficiency).12 In European guidelines, in which the majority of recommendations are based on low levels of evidence because of the rarity of UCDs, the recommended first-line medications for initial management of hyperammonemia are sodium benzoate, sodium phenylbutyrate/phenylacetate, and L-arginine. Protein intake should be minimized temporarily, but feeding needs to commence to meet metabolic demands. Following improvement of hyperammonemia (less than 100 μmol/L), reintroduction of protein and essential amino acids should not be delayed beyond 24 hours to 48 hours, increasing daily to the required amount.8
As there is no cure for UCDs, the goals for the long-term management of UCDs are to achieve normal development, to prevent hyperammonemia, and to maintain a good quality of life (QoL). These are achieved through a low-protein diet (and sometimes essential amino acids and other essential nutrients supplementation), pharmacotherapies to increase waste nitrogen excretion for patients with persistently higher ammonia levels (such as ammonia levels greater than 150 μmol/L), and liver transplantation in selected patients.4,8 Diet therapy alone is insufficient in the majority of cases, and nitrogen scavengers are usually necessary.13,14 Nitrogen scavengers used as an adjunct to diet for the long-term management of UCDs include sodium benzoate and sodium phenylbutyrate (NaPBA). In Europe, sodium benzoate is the preferred drug, whereas in North America NaPBA (Buphenyl in the US or Pheburane in Canada) is recommended as chronic maintenance therapy.8,11 Although NaPBA is considered the mainstay of pharmacological therapy in chronic management of UCDs, its use is associated with decreased appetite, taste disturbances and body odour, and it causes menstrual dysfunction/amenorrhea in one-fourth of postpubertal females.9,15 More recently, glycerol phenylbutyrate (GPB) was approved as a nitrogen-scavenging therapy.16,17 All patients should be monitored for plasma arginine and given arginine and citrulline supplementation to address impaired synthesis in the urea cycle.8 Liver transplantation is a potentially curative option for patients with UCDs, but it cannot reverse established neurologic sequelae and is associated with significant morbidities. It is recommended to be performed in patients without irreversible neurological damage who are in a stable metabolic condition, generally between three and 12 months of age.8,9
Patients with UCDs require lifelong monitoring, including anthropometric data, biochemical tests, dietary and drug review, history of intercurrent illness, and use of the emergency regimen. Visit intervals should be individualized on the basis of age, growth, severity, metabolic stability, and compliance with diet and drug therapy. Young and severely affected patients may need monitoring every three months, while annual reviews may be enough for older or less severely affected patients.8
1.3. Drug
GPB (Ravicti) is a triglyceride containing three molecules of phenylbutyric acid. Phenylacetic acid, a major metabolite of phenylbutyric acid, conjugates with glutamine through acetylation in the liver and kidneys to form phenylacetylglutamine, which is excreted by the kidneys. This provides an alternative nitrogen elimination pathway.16 After oral administration, an action of pancreatic lipases in the gastrointestinal (GI) tract is required to convert GPB into phenylacetic acid. During the absorption of NaPBA, it is rapidly metabolized to phenylacetic acid without the involvement of pancreatic lipases. Therefore, GPB acts as a slow-release form of NaPBA, achieving more stable control of ammonia levels over a 24-hour period.12,16,18 In addition, it may offer advantages with regard to tolerability and palatability, as it is a colourless and tasteless oil with no sodium content. However, its use is contraindicated in infants under two months of age because their immature pancreatic exocrine function could lead to insufficient drug metabolism.12 In March 2016, Ravicti received a Notice of Compliance by Health Canada (HC) for use as a nitrogen-binding drug for chronic management of adult and pediatric patients two years of age or older with UCDs who cannot be managed by dietary protein restriction and/or amino acid supplementation alone.17
Ravicti is available as oral liquid with 1.1 g of GPB per mL. The daily dose should be individually adjusted according to the patient’s estimated urea synthetic capacity (if any), protein tolerance, and daily dietary protein intake. An initial estimated dose for GPB during a 24-hour period is 0.6 mL/g of dietary protein ingested per 24-hour period, assuming all the waste nitrogen is covered by GPB and excreted as phenylacetylglutamine. The recommended total daily dose range of GPB is 4.5 mL/m2 per day to 11.2 mL/m2 per day (5.0 g/m2 per day to 12.4 g/m2 per day), and should be divided into equal amounts in a day, such as three times to six times per day. For patients who have been previously treated with NaPBA, the total daily dose of GPB can be calculated based on the total daily dose of NaPBA.12 Treatment with GPB must be combined with dietary protein restriction and, in some cases, dietary supplements.
View in own window
| Indication Under Review |
|---|
| As a nitrogen-binding agent for chronic management of adult and pediatric patients greater than and equal to two years of age with UCDS who cannot be managed by dietary protein restriction and/or amino acid supplementation alone. |
| Listing Criteria Requested by Sponsor |
|---|
| Per indication. |
The objective of this systematic review is to examine the beneficial and harmful effects of GPB as a nitrogen-binding drug adjunctive to dietary protein restriction and dietary supplements for chronic management of adult and pediatric (at least two years of age) patients with UCDs.
Table 2Key Characteristics of Nitrogen-Scavenging Therapies for Urea Cycle Disorders
View in own window
| GPB (Ravicti) | NaPBA (Pheburane) |
|---|
| Mechanism of action | Metabolized to release phenylbutyrate, which is then oxidized to phenylacetate. Phenylacetate conjugates with glutamine to form PAGW to be excreted by the kidneys, providing another route of nitrogen elimination. | Prodrug metabolized to phenylacetate, which conjugates with glutamine to form phenylacetylglutamine to be excreted by the kidneys, providing another route of nitrogen elimination. |
|---|
| Indicationa | For the chronic management of adult and pediatric patients ≥ 2 years of age with UCDs who cannot be managed by dietary protein restriction and/or dietary supplements. | As adjunctive therapy in the chronic management of UCDs, involving deficiencies of carbamoyl phosphate synthetase, ornithine transcarbamylase or argininesuccinate synthetase, in patients with neonatal-onset presentation and patients with late-onset disease with a history of hyperammonemic encephalopathy. |
|---|
| Route of administration | Oral (nasogastric or gastrostomy tube for patients unable to take product orally) | Oral (nasogastric or gastrostomy tube for patients unable to take product orally) |
|---|
| Dosage form | Ravicti: liquid (1.1 g/mL) | Pheburane: coated granules with 483 mg NaPBA per gram of granules |
|---|
| Recommended dose | Initial estimated dosage: 0.6 mL/24-hour period. Recommended starting dosages for patients switching from NaPBA and NaPBA-naive patients may be different.
Recommended total daily dosage: 4.5 mL/m2/day to 11.2 mL/m2/day (5.0 g/m2/day to 12.4 g/m2/day)
Daily dosage of Ravicti (mL) for patients switching from NaPBA: total daily dose of NaPBA tablets (g) × 0.86, or total daily dose of NaPBA powder (g) × 0.81.
Liquid should be divided into 3 equal doses to 6 equal doses per day.
The daily dose of Ravicti should be individually adjusted. | Patients < 20 kg: 450 mg/kg/day to 600 mg/kg/day
Patients ≥ 20 kg: 9.9 g/m2/day to 13.0 g/m2/day
The total daily dosage of Pheburane should be divided into equal amounts and given with each meal or feeding.
The daily dose of Pheburane should be individually adjusted. |
|---|
| Serious side effects/safety issues | High levels of phenylacetate may result in neurotoxicity (somnolence, fatigue, lightheadedness, etc.). | Decreased appetite, body odour, taste aversion, amenorrhea/menstrual dysfunction (females)
High levels of phenylacetate may result in neurotoxicity (somnolence, fatigue, lightheadedness, etc.). |
|---|
| Other | Not indicated for management of acute hyperammonemia. Contraindicated in patients < 2 months of age. | Should not be used for management of acute hyperammonemia. |
|---|
GPB = glycerol phenylbutyrate; NaPBA = sodium phenylbutyrate; PAGW = phenylacetylglutamine; UCD = urea cycle disorder.
- a
Health Canada indication.
Source: Health Canada product monograph for Ravicti16 and Pheburane.18