Background
Given the lack of head-to-head studies comparing ustekinumab with other relevant biologics for moderate-to-severe Crohn’s disease (CD) in this CDR review, the objective of this Appendix is to summarize and critically appraise the evidence available regarding the comparative efficacy and safety of ustekinumab versus infliximab, adalimumab, and vedolizumab through indirect comparison (IDC) using network meta-analysis (NMA) methodology. Both induction and maintenance treatment in adult patients with moderate to severely active CD were evaluated in this review. Three IDCs were assessed: the unpublished IDC submitted by the manufacturer,12 a published IDC by Mocko et al.,14,15, and another published IDC by Singh et al.13
Methods for Manufacturer’s Indirect Comparison
Study Eligibility and Selection Process
The manufacturer submitted an IDC based on a systematic review that compared ustekinumab with infliximab, adalimumab, and vedolizumab in patients with moderate-to-severe CD.12 A systematic search of randomized controlled trials was performed by searching multiple electronic databases and performing hand searches. Only biologic therapies and their respective dosages that had been approved in Canada for CD were included. Outcomes of interest for the systematic literature review included efficacy end points (Crohn’s Disease Activity Index [CDAI], C-reactive protein, fecal lactoferrin and calprotectin, mucosal healing/endoscopic improvement, fistula closure), safety end points (infections/serious infections, grade 3/4 adverse events [AEs], hospitalizations/surgery, discontinuations/withdrawals, dosage escalations), and quality of life/other measures (Inflammatory Bowel Disease Questionnaire [IBDQ], Work Productivity and Activity Impairment, Short Form [36] Health Survey [SF-36], EuroQol 5-Dimensions questionnaire). Two independent reviewers assessed titles and abstracts, and potentially relevant articles were retrieved for full-text review. Eligible articles were selected based on the inclusion and exclusion criteria provided by the manufacturer, with discrepancies resolved through discussion and/or a third reviewer.
The network analysis did not include all aforementioned outcomes but included only those that were appropriate, based on a feasibility assessment conducted at the onset of the systematic review. The feasibility assessment helped to identify trials that were considered sufficiently similar; subsequently, studies that examined certolizumab and natalizumab were eliminated, as they did not contain comparable end points, nor did they report efficacy data at one year post-treatment. In addition, neither certolizumab nor natalizumab are approved for treating Crohn’s disease in Canada. Safety end points were also not included in the NMA as they were deemed infeasible in the induction period owing to the differences in the AE definitions among trials. In addition, these safety end points were not included for the maintenance phase owing to multiple sources of heterogeneity in study design and lack of comparability among placebo arms in the various trials. Decision sets for the NMAs are included in .
Inclusion Criteria for the Manufacturer, Mocko et al., and Singh et al. Indirect Comparisons.
Quality Assessment of Included Studies
Data were extracted and quality-checked by two independent reviewers. Risk of bias was assessed using the National Institute for Health and Care Excellence (NICE) checklist, which included the following end points: adequacy of randomization and allocation concealment; similarity of prognostic factors between groups at baseline; blinding of patients, care providers, and end point assessors; similarity of dropouts; selective end point reporting; use of an intention-to-treat analysis; and adequacy of methods for handling of missing data. Results of the individual quality assessment were provided by the manufacturer.
Indirect Comparison Methods
A Bayesian hierarchical model (which helps preserve the randomization of each trial) was used for the NMA, which was performed using WinBUGS V1.4 software. The manufacturer conducted separate analyses of the subpopulation of patients who experienced treatment failure with conventional therapies, the subpopulation of patients who experienced treatment failure with anti-TNF therapies, and the overall study population (for the maintenance phase). Standard NMA methods (by Dias et al.)69 were used for the induction phase, whereas a treatment-sequence analysis was used to assess treatment efficacy at one year (induction plus maintenance). Outputs were reported as median odds ratios with corresponding 95% credible intervals (CrI), with differences considered significant if the 95% CrIs did not include the null value (1). The deviance information criterion (DIC) was used to assess the relative goodness of fit of both the fixed-effects and random-effects models, of which the model with the lowest DIC was selected for each outcome (as the better model to use for that end point). A statistical approach that adjusted to account for the correlation between treatment effects was used for trials assessing more than two treatments of interest (e.g., multi-arm trials). No assessment of inconsistency between direct and indirect evidence was performed, as there were no closed loops in the networks. Non-informative priors were used for unknown parameters. The following priors were used for the base-case analysis (BCA): normal distributions with a mean of 0 and a variance of 10,000 for treatment effects and a uniform distribution for the between-trial standard deviation, with a range of [0,2]. Convergence was assessed, and 20,000 iterations were used as a burn-in for each of the analyses. This was followed by 20,000 or more iterations for estimation and monitoring of all parameters for the fixed and random-effects models.
For the induction phase, the time points assessed were week 4 (for infliximab and adalimumab trials) and week 6 (for vedolizumab and ustekinumab trials) in order to optimize comparability between trials. The manufacturer provided a pairwise comparison for the BCA of the induction phase in the subpopulation that had experienced treatment failure with conventional therapy and the subpopulation that had experienced treatment failure with anti-TNF therapy using a fixed-effects model. This was performed in order to ascertain that results were similar to those from the NMA. In addition, numerous sensitivity analyses were conducted in order to test the robustness of the BCA results and included the following:
BCA conducted under a frequentist framework (using the Bucher method)
Different times of assessments (including week 8 results in UNITI and week 2 results from Targan et al. 1997)
Exclusion of Targan et al. 1997 from subpopulation that had experienced treatment failure with conventional therapy network
Exclusion of vedolizumab trials from the subpopulation that had experienced treatment failure with conventional therapy network
Exclusion of adalimumab trials from the subpopulation that had experienced treatment failure with anti-TNF therapy network
Exclusion of Watanabe et al. 2012 from both subpopulation networks
Inclusion of the CERTIFI trial results (included in the main body of the report) in subpopulation that had experienced treatment failure with anti-TNF therapy, as the 6 mg/kg dose in this trial was not comparable to the 6 mg/kg dose in the UNITI-1 trial
End points that selected time points based on re-randomization times.
presents details regarding the outcomes and populations evaluated in the included induction trials.
Outcomes and Populations Evaluated in Indirect Comparison for Induction Studies.
For the maintenance phase, the manufacturer used treatment-sequence analyses in which outcomes from the induction and maintenance phases were used to create an overall outcome variable. The manufacturer’s justification for the need to use this approach was first identified in its feasibility assessment of the maintenance-phase NMA. Several conceptual differences between the included trials were observed:
differences in the selection criteria required for entry into the maintenance phase
different induction active-treatment regimens for patients in the placebo arms
different assessment times
different clinical response definitions
different criteria for re-randomization
the fact that the induction phases could have been open label or double blind
whether patients continuing on to the maintenance phase had to be in remission.
Because of this heterogeneity and to minimize bias, the manufacturer decided to consider only those trials with the most comparable maintenance phases for the analysis of efficacy after one year of treatment. In addition, the manufacturer noted that placebo response rates were different and that these arms were not truly common comparators across trials. This was tested using a chi-square test that compared the observed placebo response and remission rates with the expected placebo rates if they were truly common comparators (the P value was less than 0.05 in both the populations that had experienced failures of anti-TNF and conventional therapies, thereby confirming that the placebo groups were not common comparators). Therefore, in its use of the treatment-sequence analyses, the manufacturer sought to evaluate the treatment effects over the entire treatment sequence (induction followed by maintenance for each intervention) and to increase the ability to compare placebo arms across the maintenance-phase trials.
The treatment-sequence analysis involved the incorporation of induction and maintenance data for each of the interventions. This allowed the manufacturer to obtain “relative” treatment effect estimates that took into account treatment history in the induction phase. The manufacturer multiplied the “conditional probability of maintaining response until the end of the maintenance phase”12 by the “probability of achieving response at the end of induction;”12 hence estimating the “probability of achieving and maintaining response by the end of the maintenance phase.”12 The manufacturer determined that the placebo groups were not true comparators; therefore, the placebo arm data contained imputed maintenance data (from IM-UNITI individual patient-level data) for the treatment sequence.
The manufacturer provided a pairwise comparison for the BCA of the entire treatment-sequence analysis in the subpopulations that had experienced failures of conventional therapy and of anti-TNF therapy using a fixed-effects model. This was performed in order to ascertain that results were similar to those from the NMA. In addition, numerous sensitivity analyses were conducted in order to test the robustness of the treatment-sequence analysis results and included the following:
BCA conducted under a frequentist framework (using the Bucher method)
Bayesian analysis with dosage adjustments
To replace inputs used in the analysis of the subpopulation of patients who experienced a failure with conventional therapy, inputs generated from data of patients who were truly naive to biologics from the UNITI program
Pooled maintenance doses to assess uncertainty of IDC treatment estimate effects.
presents details regarding the outcomes and populations evaluated in the included maintenance trials.
Outcomes and Populations Evaluated in Indirect Comparison – Treatment-Sequence Analysis.
Results
Study and Patient Characteristics
A total of 11 trials were included in the various NMA analyses. Only trials that assessed the efficacy of ustekinumab, adalimumab, infliximab, and vedolizumab were included. The following studies included only induction-phase results: Classic I and GAIN (adalimumab), Targan et al. 1997 (infliximab), GEMINI III (vedolizumab), and UNITI-1 and UNITI-2 (ustekinumab). The following studies included only maintenance-phase results: ACCENT I (infliximab), CHARM (adalimumab), ACCENT I (infliximab), and IM-UNITI (ustekinumab). Trials that presented both induction and maintenance results included Watanabe et al. 2012 (adalimumab) and GEMINI II (vedolizumab). Of the studies linking treatments of interest that were excluded, D’Haens 1999 (infliximab) lacked reported outcomes, Feagan 2008 (vedolizumab) used unapproved dosages, Classic II (adalimumab) included only patients in remission at weeks 4 and 8, and EXTEND (adalimumab) included a mixed population whose results were not available by subgroups. Although the CERTIFI trial (ustekinumab) was excluded from the NMA analyses (as the doses were not the same as the UNITI trials and there was a lack in reported outcomes for the 22-week analysis), it was included in a sensitivity analysis, and descriptive safety results were also reported. With regard to the included induction trials, the mean duration of CD ranged between 4.7 and 12.7 years, the mean CDAI ranged between a score of 286 and 328, the proportion of patients on corticosteroids ranged between 21.7% and 54%, and the proportion of patients on concomitant immunomodulators ranged between 16.8% and 54%. Analyses in the subpopulation who had experienced a failure of anti-TNF therapy were obtained from data in six studies (including five treatments, n = 1,433), and analyses in the subpopulation who had experienced a failure of conventional therapy were obtained from data in five studies (including six treatments, n = 1,130). Details of the induction-study characteristics are presented in .
Select Study Characteristics Included in the Indirect Comparison.
Evidence Networks
Evidence networks were provided in the manufacturer-submitted IDC. None of the networks contained any closed loops, and the networks were anchored to placebo as the only common treatment arm between studies. The following evidence networks were provided in the manufacturer’s IDC.
Induction Networks
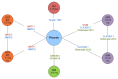
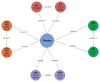
The overall network for the induction NMA is presented in The overall network for patients who had experienced a failure with conventional therapy is presented in . The overall network for patients who had experienced a failure with anti-TNF therapy is presented in .
Overall Network for the Induction Network Meta-Analysis. ADA = adalimumab; IFX = infliximab; IV = intravenous; UST = ustekinumab; VDZ = vedolizumab. Source: Manufacturer’s submitted indirect comparison.
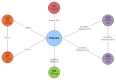
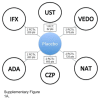
Overall Network for Patients Who Have Failed Conventional Therapy (n = 6). ADA = adalimumab; IFX = infliximab; IV = intravenous; UST = ustekinumab; VDZ = vedolizumab. Source: Manufacturer’s submitted indirect comparison.
Overall Network for Patients Who Have Failed Anti-Tumour Necrosis Factor Therapy (n = 5). Abbreviations: ADA = adalimumab; IV = intravenous; UST = ustekinumab; VDZ = vedolizumab. Source: Manufacturer’s submitted IDC.
Maintenance Phase Networks
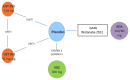
The overall network for the maintenance phase NMA is presented in . The overall network for patients who have experienced a failure with conventional therapy is presented in . The overall network for patients who have experienced a failure with anti-TNF therapy is presented in .
Overall Network for the Maintenance Phase Network Meta-Analysis in the Overall Population. ADA = adalimumab; eow = every other week; IFX = infliximab; q4w = every 4 weeks; q8w = every 8 weeks; q12w. every 12 weeks; SC = subcutaneous; UST = ustekinumab; (more...)
Overall Network for the Maintenance Phase Network Meta-Analysis in the Failed Conventional Therapy Subpopulation. ADA = adalimumab; eow = every other week; IFX = infliximab; q4w = every 4 weeks; q8w = every 8 weeks; q12w = every 12 weeks; SC = subcutaneous; (more...)
Overall Network for the Maintenance Phase Network Meta-Analysis in the Failed Anti-Tumour Necrosis Factor Therapy Subpopulation. ADA = adalimumab; SC = subcutaneous; UST = ustekinumab; VDZ = vedolizumab. Source: Manufacturer’s submitted indirect (more...)
Efficacy Results
Induction Therapy
No statistically significant differences between ustekinumab 6 mg/kg and adalimumab (either 80/40 mg or 160/80 mg) or ustekinumab 6 mg/kg and vedolizumab 300 mg were observed in either the subpopulation who had experienced a failure with conventional or anti-TNF therapy for clinical response (CDAI-70), enhanced clinical response (CDAI-100), or clinical remission (CDAI<150). Statistically significant differences in favour of infliximab 5 mg/kg compared with ustekinumab 6 mg/kg were observed for clinical response (CDAI-70) and clinical remission (CDAI < 150) in the subpopulation who had experienced a failure with failed conventional therapy (0.11; 95% CrI, 0.02 to 0.48; and 0.08; 95% CrI, 0.01 to 0.59; respectively.) Detailed NMA results for the induction period are presented in . The BCAs were determined to be robust, as the numerous aforementioned sensitivity analyses did not change the clinical interpretation (data not shown).
Network Meta-Analysis Results for the Induction Period.
Maintenance Therapy
With regard to enhanced clinical response (CDAI-100) for the maintenance phase, the only statistically significant difference was in favour of ustekinumab 90 mg every 12 weeks compared with vedolizumab 300 mg every eight weeks in the overall population (1.60; 95% CrI, 1.01 to 2.54). This statistically significant difference was not observed in either the subpopulation that had experienced a failure of conventional therapy or anti-TNF therapy. No statistically significant differences were observed in the overall population or subpopulations between the different biologic treatment regimens compared with ustekinumab 90 mg every 12 weeks with regard to clinical remission (CDAI < 150).
For the most part, no statistically significant differences between ustekinumab 90 mg every eight weeks and the other biologic regimens were observed in the overall population or subpopulations with regard to either the enhanced clinical response (CDAI-100) or clinical remission (CDAI < 150). There were two exceptions to this. First, there was a statistically significant difference in favour of ustekinumab 90 mg every eight weeks when compared with vedolizumab 300 mg every four weeks in the overall population with regard to the enhanced clinical response (CDAI-100 1.57; 95% CrI, 1.002 to 2.47). Second, there was a statistically significance difference in favour of ustekinumab 90 mg every eight weeks when compared with vedolizumab 300 mg every eight weeks in the overall population with regard to clinical remission (CDAI < 150 1.74; 95% CrI, 1.05 to 2.88). Detailed NMA results are presented in . The BCAs were determined to be robust, as the numerous aforementioned sensitivity analyses did not change the clinical interpretation (data not shown).
Network Meta-Analysis Results for the Treatment-Sequence Analysis.
Safety Results
Induction Therapy
No NMA was performed with the safety results from the induction- or maintenance-phase trials. NMA, while statistically feasible, was determined by the manufacturer to be infeasible from a clinical standpoint. That being said, safety results were reported for the induction phase as number of patients along with their respective proportions of AEs. Total AEs in all of the induction studies identified ranged from 52% to 74%, and the incidence of infections ranged from 9% to 26%. In addition, total serious AEs ranged 1% to 9%. Caution should be used when interpreting these results, as definitions of AEs differed between trials. Detailed descriptive safety results are provided in . No maintenance-phase safety results were presented.
Safety Results From the Induction Period.
Critical Appraisal of Manufacturer’s IDC
The International Society for Pharmacoeconomics and Outcomes Research (ISPOR) Task Force on Indirect Treatment Comparisons70 was used to guide the critical appraisal of the manufacturer’s submitted IDC.
The manufacturer’s rationale for conducting the IDC (i.e., absence of head-to-head studies) and the objectives of the IDC (i.e., comparisons of biologics approved for use in patients with mild-to-moderate ulcerative colitis [ustekinumab versus infliximab, adalimumab, and vedolizumab]) were clearly reported. A comprehensive systematic review was performed with a two-stage selection process, in which articles were first selected based on titles and abstracts and then full-text articles were retrieved and their inclusion criteria ascertained. In addition, data extraction was performed and quality-checked by two independent reviewers. Risk of bias was assessed using the NICE checklist (Section A1.1.2 Quality Assessment of Included Studies), and detailed results of these assessments were provided. The manufacturer provided both inclusion and exclusion criteria that were used for screening and reported lists of both included and excluded references, with accompanying reasons. Specific and detailed dosage sets were provided for both the induction and maintenance phases. The manufacturer also provided figures of all networks.
The manufacturer’s IDC described the efforts taken to assess the balance of effect modifiers across studies in order to establish the feasibility of performing an NMA. Through consultation with clinical experts, the manufacturer examined the time of assessment (which varied across trials) in the induction phase and subsequently chose the four-week time point for infliximab and adalimumab and the six-week time point for vedolizumab and ustekinumab. However, heterogeneity with regard to the timing of the assessments was still apparent in the individual studies, decreasing the confidence in the induction-phase results. In an attempt to minimize heterogeneity, the manufacturer performed separate analyses for patients who had experienced a failure with conventional therapy and with anti-TNF therapy. In addition, the manufacturer recommended caution when interpreting results of ustekinumab compared with infliximab for the induction phase for all outcomes, as the only study included in the IDC containing infliximab was an older and smaller study (ACCENT I), in which there were numerous issues (e.g., missing and non-evaluable placebo arm data, smaller magnitude of effects with the higher versus lower doses of infliximab, and lack of result reproducibility in the open-label induction phase). It should also be noted that there were some differences between the baseline patient characteristics (especially in regard to the ACCENT I infliximab trial), such as the mean duration of disease, mean CDAI score, C-reactive protein at baseline, smoking status, IBDQ scores, and disease phenotype. This further increased the uncertainty, decreased confidence in the results, and decreased the generalizability of both the induction and treatment-sequence analysis results for patients with moderate-to-severe CD.
With regard to the maintenance phase, the manufacturer undertook a feasibility assessment and noted that the similarity assumption had been violated as a result of the variation among studies in the selection criteria for entry into maintenance treatment. The feasibility assessment also noted that the transitivity assumption may have been violated, as the manufacturer identified different placebo rates. This aspect led the manufacturer to believe that the placebo arms across trials were not true comparators (clinical heterogeneity). Statistical heterogeneity was assessed using a chi-square test for remission and response in the subpopulations who had experienced a failure with conventional and anti-TNF therapies, showing P values less than 0.05, indicating heterogeneity. The manufacturer analyzed the maintenance of efficacy after one year using only those trials with the most comparable maintenance phases and performing a treatment-sequence analysis, in which subgroup data were available for results from the induction phases of these trials. It should be noted that all the placebo arms corresponded to patients who received only active induction therapy. In addition, the exclusion of trials that did not include the most comparable maintenance phase for the analysis of efficacy after one year of treatment (the manufacturer did not provide a list of excluded trials) may introduce a risk of bias associated with the loss of applicable information. The treatment-sequence analysis method includes both induction and maintenance data for each intervention. In addition, imputed data (from individual patient-level data from IM-UNITI) was used for the maintenance data in the placebo arms, and weighted averages for the maintenance placebo rates were used for the maintenance placebo-to-placebo arms. However, this methodology is exploratory, as this method has not been validated. Data were imputed for the placebo arms and carries with it some uncertainty. Further, the approach is problematic because it does not maintain randomization past the induction phase. Therefore, caution must be used when interpreting the results because of the potential for bias and confounding, which have not been controlled for.
In order to assess the robustness of the BCA NMA results, numerous sensitivity analyses were conducted (Section A1.1.2 Indirect Comparison Methods). The results of the sensitivity analyses for all of the outcomes and populations (or subpopulations) indicated that all of the BCAs were robust. In addition, the manufacturer performed pairwise comparisons for the BCAs in the induction phase and complete treatment-sequence analysis for the two differing subpopulations (who had experienced a failure with conventional and anti-TNF therapies) to ensure that the NMA results were robust (which they were).
In order to observe model fit, DICs were calculated using both the fixed-effects and random-effects model for each outcome (CDAI-70, CDAI-100, and CDAI < 150), for each subpopulation (who had experienced a failure with conventional therapy or with anti-TNF therapy), and for both the induction phase and the treatment-sequence analyses. The model with the lowest DIC was the one used, with the fixed-effects model consistently favoured for every analysis.
The reviewers extracted data for more outcomes than were assessed in the NMA, but only three outcomes (clinical response [CDAI-70], enhanced clinical response [CDAI-100], and clinical remission [CDAI < 150]) were included. Although the CDAI is a validated measure of disease activity in CD and is composed of many separate outcomes (number of liquid stools, abdominal pain, well-being, abdominal mass, extra-intestinal manifestations, use of antidiarrheal drugs, body weight, and hematocrit values), other outcomes also help to inform how effective treatment is. In addition, while the trials themselves were probably not powered to detect statistically significant changes in the secondary outcomes, comparisons of outcomes such as mucosal, histologic, and endoscopic healing (which often correspond with clinical remission and clinical response), anemia, and patient-reported outcomes would still have been informative, as long as methodological issues were identified and mentioned. It should also be noted that, although statistically significant differences were observed in favour of infliximab 5 mg/kg when compared with ustekinumab 6 mg/kg for clinical remission (CDAI < 150) in the induction phase, one should remain skeptical that clinical remission is possible at such an early time point, as the clinical expert who was consulted on this submission pointed out.
All of the included randomized controlled trials (RCTs) were placebo-controlled, and none contained any head-to-head comparisons of the relevant biologic drugs. For this reason, none of the networks contained closed loops with regard to the biologics, precluding the ability to assess consistency between direct and indirect comparisons. Hence, the NMA would have been stronger if there were a closed loop (e.g., active comparison). In addition, all of the trials were, at most, one year long; hence, longer-term efficacy and safety were not assessed.
Safety outcomes were not included in any NMA for either the induction or maintenance phases. The manufacturer determined that conducting analyses on subpopulations would lead to small numbers of events being assessed, a loss of statistical power, and a lack of model convergence. It also noted that using a pooled/mixed population would be unacceptable from a clinical standpoint as a result of confounding. In addition, it noted that many AEs included gastrointestinal events that are related to CD, potentially introducing uncertainty about true treatment effects. This being said, an NMA increases the power of the assessment of AEs, and performing an NMA on either AEs or SAEs in these subpopulations would still provide insight into how patients with moderate-to-severe CD are adversely affected by the use of these different biologics.
Methods for Mocko et al
Study Eligibility and Selection Process
Mocko et al.14,15 conducted an IDC, based on a systematic review of the literature, that compared and evaluated the safety of biologic drugs (adalimumab, infliximab, certolizumab pegol, ustekinumab, and vedolizumab) with one another or with placebo in patients with CD (defined by conventional radiographic, endoscopic, and clinical criteria [CDAI > 150]). The biologics included had to have been approved for the treatment of CD by either the European Medicines Agency (EMA) or FDA; except ustekinumab which, at the time of this IDC, was undergoing the approval process for patients with CD by the EMA and FDA. Inclusion criteria for this IDC are outlined in . A systematic search of randomized controlled trials published in English was performed using multiple databases. Studies were included if the investigators examined the induction phase (follow-up of six to 10 weeks) or maintenance phase (follow-up of 52 to 56 weeks). Two independent reviewers assessed titles, abstracts, and full-text articles for inclusion in the IDC, with any discrepancies resolved by consensus with a third reviewer. All safety outcomes with a frequency of at least 3% were included and extracted by the same two independent reviewers. Only data from studies that adhered to the approved dosage regimens (approved by the EMA or FDA) were included in the IDC, and results for the induction and maintenance phases were analyzed separately. Selected trial characteristics are provided in .
Selected Study Characteristics Included in the Mocko et al. Indirect Comparison.
Quality Assessment of Included Studies
The same two independent reviewers evaluated study quality using a domain-based evaluation tool recommended by the Cochrane Collaboration, and any ambiguities were resolved through consensus with a third reviewer. The domain-based tool examined the following domains: random sequence generation, allocation concealment, blinding (participants and investigators), incomplete outcome data, selective reporting, and other bias. Results of the individual quality assessments were provided by the manufacturer.
Indirect Comparison Methods
A Bayesian random-effects model (using the Markov chain Monte Carlo simulation) was used for the NMA, which was performed using the Aggregate Data Drug Information System (ADDIS) software, version 2. A consistency model was used, and this was based on 20,000 iterations for each of the four chains with a 5,000 iteration burn-in period. The assessment of convergence was performed using the Brooks–Gelman–Rubin method. Outcomes were expressed as median odds ratios with corresponding 95% CrIs. As previously stated, results for the induction and maintenance phases were analyzed separately. Clinical heterogeneity was assessed by the extraction and examination of the following trial characteristics: study design, dosage, treatment duration, follow-up, patients achieving predefined outcomes.
A random-effects pairwise meta-analysis was performed in order to obtain direct estimates of effects relative to placebo, which were used to confirm the results obtained in the NMA framework. Estimates of effects were presented as odds ratios and 95% confidence intervals (CIs). Heterogeneity for the pairwise meta-analyses was planned to be assessed using the I2 parameter (with values of 0% representing no heterogeneity and those higher than 50% indicating significant heterogeneity). However, no formal assessment of statistical heterogeneity was performed as a result of the limited number of RCTs for the pairwise comparisons. In the absence of an assessment of statistical heterogeneity, the authors used a conservative random-effects model with statistical significance defined as a P value less than 0.05.
Results
Study and Patient Characteristics
Ten RCTs were included in the NMA analyses. Of these studies, three were on the use of adalimumab (CHARM, CLASSIC II, and Watanabe et al.), one was on the use of certolizumab pegol (Sandborn et al.), three were on the use of infliximab (ACCENT I, ACCENT II, and Regueiro et al.), one was on the use of ustekinumab (CERTIFI), and two were on the use of vedolizumab (GEMINI II and GEMINI III). With regard to the induction phase, Watanabe et al. (adalimumab), Sandborn et al. (certolizumab pegol), GEMINI II and GEMINI III (vedolizumab), and CERTIFI (ustekinumab) were considered. For the maintenance phase, Watanabe et al., CHARM, and CLASSICII (adalimumab), ACCENT I, ACCENT II, and Regueiro et al. (infliximab), and GEMINI II (vedolizumab) were considered. Using placebo as the common comparator, Mocko et al. determined that there were no significant differences among these groups (based on their characteristics); hence, they used the placebo group as the link for the NMA. No statistical heterogeneity was observed for the induction phase (I2 = 0%) for all analyzed outcomes, and the same lack of heterogeneity was observed in the maintenance phase for the majority of the outcomes. The only exceptions to this in the maintenance phase were, in the use of infliximab, injection-site reactions and AEs leading to study drug discontinuation (I2 = 70% to 86%) and, in the use of adalimumab, infections and nasopharyngitis (I2 = 73% to 80%). Convergences were achieved at 20,000 simulations for all of the end points. All of the included RCTs were evaluated as having a high risk of bias, especially with regard to the “incomplete outcome data” domain. In addition, there was an unclear risk of bias regarding “allocation concealment” in six RCTs, whereas there was an apparent low risk of bias in the other domains. Specific patient characteristics for the individual trials (e.g., age, sex, mean duration of CD, proportion of patients who are treatment-naive versus -experienced, concomitant medications, etc.) were not provided.
Evidence Networks
Evidence networks were provided by Mocko et al. None of the networks contained any closed loops, and networks were anchored to placebo as the only common treatment arm between studies. The following evidence networks were provided.
Induction and Maintenance Networks
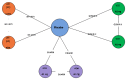
The overall network for the induction NMA is presented in . The overall network for the maintenance NMA is presented in .
Overall Networks for the Induction and Maintenance Phases for Mocko et al. Source: Reprinted from Pharmacological Reports, 68/6, Mocko P, Kawalec P, Pilc A, Safety profile of biologic drugs in the therapy of Crohn disease: a systematic review and network (more...)
Safety Results
Induction Therapy
No statistically significant differences were evident among any of the biologics examined in the induction-phase NMA with regard to any AEs, infections, injection-site reactions, SAEs, and drug discontinuations due to AEs. In addition, no statistically significant differences were observed among biologics for individual AEs, such as abdominal pain, arthralgia, headache, nausea, or nasopharyngitis. The pairwise meta-analyses against placebo echoed the lack of statistical significance (data not shown). Detailed NMA results for the induction phase are provided in .
Network Meta-Analysis Safety Results for the Induction Period.
Maintenance Therapy
No statistically significant differences were evident among any of the biologics (adalimumab, infliximab, or vedolizumab) examined in the maintenance-phase NMA with regard to any AEs, infections, serious infections, injection-site reactions, SAEs, and drug discontinuations due to AEs. In addition, no statistically significant differences were observed among any of the aforementioned biologics for the individual AEs, such as abdominal pain, arthralgia, headache, nausea, nasopharyngitis, pyrexia, or upper respiratory infections. In contrast to this, a statistically significant difference in favour of adalimumab over placebo was observed for injection-site reactions and in favour of placebo over vedolizumab for any AEs and SAEs (data not shown). No comparisons were available for ustekinumab in the maintenance phase.
Critical Appraisal of Mocko et al. Indirect Comparison
While the IDC by Mocko et al. included acceptable methods in terms of the systematic review portion, some major limitations preclude definitive conclusions with regard to the results.
The rationale and objectives for conducting the IDC on the safety of these biologics for patients with CD were clearly reported, as were the methods for the systematic review and their statistical analyses. A comprehensive systematic review was performed with a two-stage selection process: articles were first selected based on titles and abstracts and then full-text articles were retrieved and their inclusion criteria ascertained. In addition, data extraction was performed and quality-checked by two independent reviewers. The two independent reviewers also assessed the risk of bias using a tool recommended by the Cochrane Collaboration based on the aforementioned domains (reported in Section A1.2.1 Quality Assessment of Included Studies). In addition, the authors were not funded externally by any pharmaceutical company, and no conflicts of interest were reported.
Mocko et al. did not report (either in the main citation or supplemental information) the individual trial or patient characteristics for the studies included in their IDC. This lack of reporting adds to the uncertainty regarding not only the severity of the disease, but also the heterogeneity of the patients across studies (although most of the studies, but not all, were included in the other IDCs). The authors also did not specify that all patients must have moderate-to-severe CD for inclusion in the IDC; instead, patients were included if they had clinical, endoscopic, and radiologic evidence of CD and CDAI greater than 150. While this is consistent with some of the other IDCs,12,13 this score does not definitively place an individual in the moderate-to-severe category of CD. For instance, according to international definitions of CD based on the CDAI parameters, scores above 150 can be classified as mild-to-moderate (CDAI 150 to 220), moderate-to-severe (CADI 220 to 450), or severe/fulminant (CDAI > 450).71 Other factors (such as age, mean duration of CD, concomitant medications, individual baseline CDAI, and other clinical, endoscopic, or radiologic assessments, etc.) were not reported; therefore, external readers are unable to ascertain the baseline heterogeneity of the patients in the included studies. There was also no mention of whether the patients were treatment-naive or treatment-experienced and no mention of any subgroup analyses (a priori or post hoc) that would address these potential confounders. This lack of reporting also extended to information on whether the authors assessed both the random- and fixed-effects models (and subsequent DIC calculations to ascertain the model of best fit) and which priors were used. All of this lack of reporting furthers the uncertainty surrounding the effect estimates that were obtained.
The authors made the assumption that there were no significant differences among patients in the placebo groups and therefore used the placebo groups as the common comparator and anchor for the networks. However, differences in the placebo groups, especially in those in the maintenance phase (as previously discussed in the manufacturer’s submitted IDC12) preclude simply “using” the placebo group as a common comparator, although this is often the main anchor in the NMA process. This adds additional uncertainty with regard to the safety outcomes assessed. In addition, there were no closed loops in either the induction- or maintenance-phase networks, precluding any assessment of consistency.
The authors performed an assessment of bias in the included studies, noting a high risk of bias regarding incomplete outcome date (or attrition bias) in all 10 RCTs and an unclear risk of bias regarding allocation concealment (or selection bias) in six RCTs. Since Mocko et al. looked only at safety outcomes with an incidence of at least 3%, this high risk of bias associated with the incomplete outcome data further increases the uncertainty associated with the outcome results. Hence, results need to be interpreted with caution. In addition, the full safety profile of these biologics was not ascertained (as AEs with an incidence of less than 3% were excluded); therefore, one must keep these results in context.
The authors noted that the limited length of the follow-up period was a significant limitation for assessment of all possible AEs, particularly with regard to the induction phase of up to 10 weeks for certolizumab pegol, infliximab, and ustekinumab. While the maintenance-phase analysis did extend the timelines, AEs or SAEs such as cancer may not have been observed during this longer follow-up period (up to 54 weeks), and a longer length of follow-up would have been more appropriate. That being said, this is a flaw of the RCTs themselves, rather than of the NMA. It should also be noted that there was no safety evidence for ustekinumab following the induction phase; therefore, no conclusions outside of the induction phase can be made.
Finally, most RCTs are designed with the power to detect efficacy end points as their primary and secondary end points; however, the same cannot be said of the safety end points. Although an NMA allows for an assessment of these types of outcomes because of the larger sample size (using multiple RCTs), the absence of a sufficiently large sample of patients to detect less commonly observed safety outcomes in the original RCTs contributes to the uncertainty surrounding the NMA results obtained.
Methods for Singh et al
Study Eligibility and Selection Process
Singh et al.13 conducted an IDC that compared and evaluated the relative efficacy of biologic drugs (adalimumab, certolizumab pegol, infliximab, natalizumab, ustekinumab, and vedolizumab) with one another or with placebo in treatment-naive (biologic-naive) patients with moderate-to-severe CD (defined on a CDAI greater than 220 but less than 450). Inclusion criteria for this IDC are outlined in . A systematic search of RCTs (with no language restriction) was performed using multiple databases. For trials assessing induction therapy, only those that assessed outcomes in patients not previously exposed to biologics or that reported results separately for patients not previously treated with biologics were included. Similarly for the maintenance phase, trials were excluded when outcomes were not reported separately for patients not previously treated with biologics in those who initially responded to induction therapy. Two independent reviewers assessed titles, abstracts, and full-text articles for inclusion in the IDC, with conflicts resolved by consensus only when examining the full-text articles. In addition, bibliographies of the included studies were searched, along with abstracts from major gastroenterology conferences, in order to obtain unpublished information. The primary outcome of interest was the induction of clinical remission in biologic-naive patients with active CD (up to 14 weeks) and the maintenance of remission of the patients who initially responded to biologics in the induction phase (up to 60 weeks). A hierarchical approach was used in both the induction and maintenance phases, in which CDAI < 150 was preferred, followed by CR-100 (failure to achieve a reduction of more than 100 points from baseline), followed by CR-70 (failure to achieve a reduction of 70 points from baseline). Selected trial and patient characteristics are provided in .
Selected Study Characteristics Included in the Singh et al. Indirect Comparison.
Quality Assessment of Included Studies
Two independent reviewers evaluated the quality of the included studies using criteria set out by the Evidence-Based Gastroenterology Steering Group. The criteria included random allocation concealment, patient and caregiver blinding, equal co-intervention use for both the placebo and treatment arms, follow-up of study patients (completeness), and the use of an intention-to-treat analysis.
Indirect Comparison Methods
A Bayesian random-effects model (using the Markov chain Monte Carlo simulation) was used for the NMA, which was performed using WinBUGS 1.4.3. The comparative efficacy between two treatments was assessed as a function of the active treatment relative to the reference (placebo) treatment. Non-informative priors were used, along with a burn-in of 10,000 iterations. The Markov chain Monte Carlo model used 100,000 simulations. Results were reported as a relative risk (RR) with 95% CrIs, and the authors adjusted for trials with multiple arms.
Active drugs and placebo were also compared in pairwise analyses using a random-effects model, with results presented as a pooled RR with 95% CIs. Statistical heterogeneity (although not formal assessment of clinical heterogeneity) was assessed using the I2 statistic, and publication bias was examined using Egger’s regression test to produce funnel-plot symmetry.
Results
Study and Patient Characteristics
Seventeen RCTs were included in the NMA, with 11 RCTs focused on induction of remission and nine trials focused on the maintenance of remission. With regard to the induction trials, adalimumab, certolizumab pegol, infliximab, natalizumab, and vedolizumab were each assessed in two trials, whereas only one study assessed ustekinumab. There was variability in the proportion of patients taking concomitant immunosuppressive drugs (range of 4% to 100%), corticosteroids (range of 21% to 100%), and 5-ASA drugs (range of 37% to 100%) between trials. With regard to the maintenance trials, three studies assessed adalimumab; one study assessed certolizumab pegol; two studies assessed infliximab; and one study assessed each of natalizumab, ustekinumab, and vedolizumab. The percentage of patients receiving concomitant medications ranged from 0% to 44% (immunomodulators), 12% to 56% (corticosteroids), and 41% to 100% (5-ASAs) in those trials that reported them. Based on the assessment of the risk of bias for the induction studies, one study failed to report whether the random allocation was concealed, one study did not report on the equal use of co-interventions in both treatment and placebo arms, and one study did not report on the use of the intention-to-treat analysis. With regard to the assessment of the risk of bias for the maintenance studies, four studies failed to report on the equal use of co-interventions in both treatment and placebo arms.
Evidence Networks
Evidence networks were provided by Singh et al. None of the networks contained any closed loops, and networks were anchored to placebo as the only common treatment arm between studies. The following evidence networks were provided.
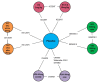
Induction Network
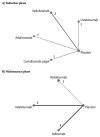
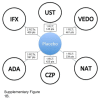
The overall network for the induction NMA is presented in .
Overall Networks for the Induction Phase for Singh et al. ADA = adalimumab; CZP = certolizumab pegol; IFX = infliximab; NAT = natalizumab; RCT = randomized controlled trial; VEDO = vedolizumab; UST = ustekinumab. Source: Reprinted from Mayo Clinic Proceedings, (more...)
Maintenance Network
The overall network for the maintenance NMA is presented in .
Overall Networks for the Maintenance Phase for Singh et al. ADA = adalimumab; CZP = certolizumab pegol; IFX = infliximab; NAT = natalizumab; RCT = randomized controlled trial; VEDO = vedolizumab; UST = ustekinumab. Source: Reprinted from Mayo Clinic Proceedings, (more...)
Efficacy Results
Induction Therapy
When ustekinumab (RR 0.1; 95% CrI, 0.02 to 0.52), vedolizumab (RR 0.23; 95% CrI, 0.06 to 0.78), natalizumab (RR 0.22; 95% CrI 0.06 to 0.70), and certolizumab pegol (RR 0.24; 95% CrI, 0.07 to 0.73) were compared with infliximab, there was a statistically significant difference in favour of infliximab for the induction of remission. No other differences were observed with regard to the induction of remission. Detailed NMA results for the induction period are provided in . The anti-TNF drugs were superior to placebo in the standard pairwise meta-analysis, while the anti-integrins and the IL-12/IL-23 antogonist (ustekinumab) were not superior to placebo for the induction of remission (data not shown).
Network Meta-Analysis Results in Biologic-Naive Patients in the Singh et al. Indirect Comparison.
Maintenance Therapy
No statistically significant differences among any of the biologics were evident for maintaining remission in those patients who were biologic-naive upon entering the induction phase and were subsequent responders in the induction phase. Detailed results for the maintenance period are provided in . With regard to the standard pairwise meta-analysis for the maintenance of remission, anti-TNF drugs and the IL-12/IL-23 antagonist were statistically significantly superior when compared with placebo; however, the same was not observed with the anti-integrins (data not shown).
Critical Appraisal of Singh et al. Indirect Comparison
The IDC by Singh et al. appeared to use acceptable methods for the systematic review; however, there are some major limitations which lessen confidence in the results.
The authors’ rationale and objectives for conducting the IDC on the efficacy the aforementioned biologics for inducing and maintaining remission in biologic-naive patients were clearly reported, as were the methods for the systematic review and their statistical analyses. In describing the NMA process, the authors did report that they adjusted for trials with multiple arms; however, details on this adjustment were lacking. A comprehensive systematic review was performed, in which articles were first selected based on titles and abstracts, and then full-text articles were retrieved and their inclusion criteria ascertained. However, there was uncertainty surrounding whether the dual selection pertained to both the title and abstract selection and full-text article selection, as it was described solely for the latter. If the selection pertained solely to the full-text articles (instead of being reported only for full-text articles), then certain articles may have been missed. Data extraction was performed and quality-checked by two independent reviewers, as was the assessment of bias. However, the criteria assessed were less stringent than those of the other two IDCs;12,14,15 the authors did not assess the similarity of prognostic factors between groups at baseline (no assessment of clinical heterogeneity), selective end point reporting, or the adequacy of methods for handling missing data. Although they assessed both patient and caregiver blinding, they did not appear to assess the blinding of the end point assessor. Some of the authors had stated conflicts of interest, and the NMA was funded by a grant from the Kern Center for the Science of Health Care Delivery, which is part of the Mayo Clinic.
In order to obtain efficacy results solely for the population of biologic-naive patients, the authors included only data on this subgroup for induction therapy. In addition, they subsequently included only data from the subset of patients who responded to induction therapy for the efficacy assessment in the maintenance phase. Induction trials that did not report outcomes separately for biologic-naive patients were excluded. In addition, maintenance trials that did not report outcomes separately for the subset of patients who responded to induction therapy were also excluded. In addition, by including only the subset of patients who responded to induction therapy in the maintenance trials, the authors artificially enriched the population of patients with those who were more likely to see an increased benefit on these biologics for the maintenance of remission. This further increases the uncertainty with regard to the results and decreases the generalizability of this treatment for those who are biologic-naive.
As with all NMAs, the primary objective was to compare drugs across similar populations of patients with predominantly the same characteristics. Although this IDC attempted to do that, there were inherent differences within the populations (as observed in ); for example, some prognostic factors were different (e.g., disease duration, use of concomitant immunomodulatory medications, phenotype of disease). In addition, there were differences in the assessment timing for both the induction and maintenance phases among the included RCTs, which could have led to differences in responses. These issues, along with the fact that there were no closed loops in either the induction- or maintenance-phase networks, also call into question the generalizability of the results and further undermine confidence in them.
As in the manufacturer’s submitted IDC, the lack of other end points (e.g., mucosal healing) does not allow one to obtain the full picture of efficacy for this class of drugs. As the clinical expert consulted for this review mentioned, although remission is a good overall end point, it is usually unattainable during the induction phase and does not fully encompass the entire realm of a successful treatment. Endoscopic evidence of remission (which is not always feasible to obtain in every patient), including mucosal healing, is an important efficacy end point that should be looked at.
This IDC did not attempt to perform an NMA on any safety end points. Although most RCTs are not powered to detect differences in safety end points, it is still beneficial (especially considering the classes of drugs involved with the treatment of CD) to attempt an NMA of safety end points. This would have provided some insight into the safety aspects involved in treating patients with moderate-to-severe CD with these biologics.
Conclusions
The manufacturer submitted an IDC12 of ustekinumab versus infliximab, adalimumab, and vedolizumab using Bayesian NMA with placebo as the common comparator (for the induction phase) and using treatment-sequence analyses (which includes induction and maintenance data) for the maintenance phase. The manufacturer noted that there were no statistically significant differences in clinical response (CDAI-70), enhanced clinical response (CDAI-100), or clinical remission (CDAI < 150) among ustekinumab 6 mg/kg and either adalimumab 80/40 mg, adalimumab 160/80 mg, or vedolizumab 300 mg in the induction phase in either the subpopulation who had experienced a failure with conventional and anti-TNF therapies. Statistically significant differences in clinical response (CDAI-70) and clinical remission (CDAI < 150) in favour of infliximab 5 mg/kg compared with ustekinumab 6 mg/kg were apparent in the subpopulation that had experienced failure with conventional therapy. However, the degree of heterogeneity noted for the network comparisons, including infliximab, reduced the certainty around these results.
For treatment-sequence NMA results (based on an exploratory methodology; hence, results should be interpreted with caution), the only statistically significant differences observed were in favour of ustekinumab 90 mg every 12 weeks when compared with vedolizumab 300 mg every eight weeks when looking at enhanced clinical response (CDAI-100) in the overall population (results were not statistically significantly different among the subpopulations). All other results showed no differences between ustekinumab 90 mg every 12 weeks and the other biologic treatment regimens. With regard to ustekinumab 90 mg every eight weeks, statistical significance in favour of ustekinumab was evident over vedolizumab 300 mg every four weeks for enhanced clinical response (CDAI-100) in the overall population (however, not in the subpopulation results) and over vedolizumab 300 mg every eight weeks for clinical remission (CDAI < 150) in the overall population (however, not in the subpopulation results). Given the uncertainty in the treatment-sequence analysis methodology and heterogeneity across studies, the comparative efficacy of these drugs in the maintenance phase of treatment is highly uncertain.
Mocko et al.14,15 reported that there were no statistically significant differences in the incidence of AEs, serious AEs, discontinuations due to AEs, or for some of the more prominent AEs (e.g., infections, injections site reactions, nausea, headache, arthralgia, etc.) among adalimumab, ustekinumab, or vedolizumab during induction therapy and among adalimumab, infliximab, and vedolizumab during maintenance therapy in patients with CD. However, some of the major limitations associated with this IDC (e.g., lack of reporting of individual trial and patient characteristics for included studies, inclusion of patients with CDAI > 150 [with no further partitioning], lack of subgroup analyses, the bias assessment of the individual studies, lack of full safety profile [including only AEs with a frequency of 3% or greater], etc.) introduce uncertainty regarding the NMA results, decrease confidence in the results, and decrease the generalizability of the results to those patients with moderate-to-severe CD. Hence, caution is required when interpreting the authors’ observations that there are no differences in safety among these drugs during the induction and maintenance phases of therapy for patients with CD.
The authors of the Singh et al.13 IDC reported a statistically significant difference in favour of infliximab when compared with the other biologic drugs they assessed (adalimumab, ustekinumab, and vedolizumab) for the induction of remission in biologic-naive patients with moderate-to-severe CD. The results of the IDC indicated that there were no statistically significant differences among these drugs for maintaining remission in biologic-naive patients with moderate-to-severe CD. However, the limitations associated with this IDC (e.g., less stringent assessment of included study biases, the exclusion of studies in which results were not separated for the biologic-naive and biologic-experienced patients [thereby potentially losing applicable evidence], the enrichment of patients more likely to respond to biologic therapy in the maintenance phase, differences in the prognostic factors associated with the baseline patient characteristics of the individual RCTs, etc.) decrease confidence in the NMA results, decrease the generalizability of the results to those patients who are biologic-naive, and increase uncertainty. Hence, one must use caution when interpreting and using these results.