The clinical evidence included in this review of icosapent ethyl is presented in three sections. Section 1, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH Common Drug Review (CDR) and Health Canada, as well as those studies that were selected according to an a priori protocol. Section 2 includes indirect evidence from the sponsor (if submitted) and indirect evidence selected from the literature that met the selection criteria specified in the review. Section 3 includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Findings From the Literature
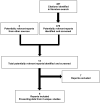
A total of two studies were identified from the literature for inclusion in the systematic review (). The included studies are summarized in . A list of excluded studies is presented in Appendix 2.
Flow Diagram for Inclusion and Exclusion of Studies.
Details of Included Studies.
Description of Studies
Two double-blind, randomized, placebo-controlled trials met the inclusion criteria for the review: the ANCHOR26 and the REDUCE-IT25 studies.
The ANCHOR trial was conducted from December 2009 to February 2011 across 97 centres in the US. The study aimed to evaluate the difference in the percent change in TG levels from baseline to week 12 for icosapent ethyl (2 g and 4 g) compared to placebo in patients with high risk for CVD and with TG levels ≥ 2.3 mmol/L (200 mg/dL) and < 5.6 mmol/L (500 mg/dL). The investigators screened 2,309 patients for eligibility, of which 1,602 patients were considered screening failures, mainly because they did not satisfy the inclusion criteria (n = 1,461) or because they withdrew consent (n = 102). The screening period consisted of four to six weeks with diet and lifestyle stabilization, after which a 12-week double-blind period ensued. After randomization procedures, investigators distributed 702 patients to three arms of study: an intervention arm with icosapent ethyl at 4 g per day (n = 233), a second intervention arm using icosapent ethyl at 2 g per day (n = 236), and a placebo group (n = 233). The 2 g per day regimen is not included in this review because it was not a dosage submitted to Health Canada. The randomization list was stratified according to the type of statin (atorvastatin, rosuvastatin, or simvastatin), presence of diabetes, and gender.
The REDUCE-IT study was a larger randomized trial evaluating clinical outcomes in addition to TG blood levels and harms. It was conducted between November 2011 to August 2016 in 473 centres from 11 countries. The screening (run-in) period consisted of one month of assessment for eligibility, after which patients were randomized 1:1 to 4 g per day of icosapent ethyl or placebo. Randomization was stratified by CV risk category, use of ezetimibe, and geographical region (a group of western countries, Eastern European countries, and the Asia–Pacific region). Of 19,212 screened patients, 8,179 were randomized to intervention or control groups. Most of the screening failures were because the patients did not meet the inclusion criteria (n = 10,429), withdrew consent (n = 340), or were lost to follow-up during this period (n = 108). The median follow-up time was 4.9 years and up to 6.2 years.
Populations
Inclusion and Exclusion Criteria
The ANCHOR study included patients older than 18 years of age with BMI 45 kg/m2 or lower, fasting TG levels of 2.3 mmol/L (200 mg/dL) or higher and less than 5.6 mmol/L (500 mg/dL), a stable dose of statin therapy (with or without ezetimibe), and at high risk for CVD. Patients at high risk for CVD were defined as having clinical coronary heart disease or clinical coronary heart disease risk equivalents (10-year risk ≥ 20%), as delineated in the National Cholesterol Education Program Adult Treatment Panel III guidelines; i.e., when one of the following were present: history of coronary artery disease (MI, angina, coronary procedure), or atherosclerotic disease (e.g., peripheral artery disease, transient ischemic attack, carotid obstruction). Patients were excluded if they were receiving non-study lipid-altering medications or other statins not stated in the protocol; if they had hemoglobin A1C greater than 9.5%; or if they had had percutaneous coronary intervention within four weeks before screening, hospitalization within four weeks before screening, known nephrotic proteinuria, or other major conditions.
The REDUCE-IT trial included patients who were 45 years of age and older and had established risk for CVD (secondary prevention), or who were 50 years of age and older and were considered at high risk (primary prevention), defined as having diabetes plus one additional risk factor for CVD. Both groups also had to have a fasting TG level of 1.7 mmol/L (150 mg/dL) or greater and less than 5.6 mmol/L (500 mg/dL), and LDL-c greater than 1.0 mmol/L (40 mg/dL) and less than 2.6 mmol/L (100 mg/dL) and had to be on stable statin therapy.
Patients that investigators categorized as high risk in the ANCHOR study would have been classified as in the established-risk (secondary prevention) group in the REDUCE-IT trial. For this reason, we did not consider the ANCHOR trial to have high-risk patients, but rather all were managed as established CVD (secondary prevention) patients ().
Summary of Baseline Characteristics — ANCHOR Study.
Baseline Characteristics
Baseline characteristics were considered similar between the intervention and placebo groups in both studies, denoting a randomization process that produced an appropriate balance of known or unknown prognostic factors, baseline conditions, medications, or prior treatments ( and ). Statin intensity was defined according to the American College of Cardiology/American Heart Association (ACC/AHA) Blood Cholesterol Guidelines (Appendix 4).
Summary of Baseline Characteristics — REDUCE-IT Study.
Interventions
In both studies, the intervention groups received icosapent ethyl as 1 g liquid-filled, oblong, gelatin capsules, at a dosage of 4 g per day. The placebo groups differed between these two studies. In the REDUCE-IT study, the placebo used was mineral oil in 1 g capsules administered as two capsules twice daily taken with food or about 4 mL per day (4 g per day). The ANCHOR study had a different approach by using a three-arm design: the control group, using four capsules filled with liquid paraffin as placebo administered twice a day orally, an icosapent ethyl 4 g daily group and an icosapent ethyl 2 g daily group.
Certain concurrent medications were permitted in both studies. Because both studies included patients with persistent hypertriglyceridemia, clinicians were allowed to administer statins as long as they were used according to the protocol. In the ANCHOR study, for instance, investigators were allowed to change patients from a non-study statin to one allowed by the protocol, such as atorvastatin, rosuvastatin, or simvastatin. The choice of statin was left to the discretion of the investigator/clinician. Other non-statin lipid-altering medications, as well as corticosteroids, weight-reduction agents, protease inhibitors to treat HIV, cyclophosphamide, or isotretinoin, were not permitted. No difference was noted between groups in the proportion of patients that used antihypertensive drugs, antidiabetic medications, statins, or other co-interventions.
Meanwhile, in the REDUCE-IT trial, non-statin lipid-altering medications or supplements were prohibited during the duration of the study, including niacin, fibrates, prescription omega-3 fatty acid medications, dietary supplements having omega-3 fatty acids (e.g., flaxseed, fish, krill, or algal oils), bile acid sequestrants, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, cyclophosphamide, and systemic retinoids. Use of any of these products during the study had to be “for compelling medical reasons” and documented. Statins, ezetimibe, herbal products, and dietary supplements not containing omega-3 fatty acids were allowed and were balanced between study groups. Modifications in drug choice and dosages on these co-interventions or additions of other medications were allowed “for compelling medical reasons” and left to the discretion of the investigator/physician.
Outcomes
The primary efficacy outcome in the ANCHOR trial was the percent change in TG level from baseline to 12 weeks. The pre-specified secondary efficacy outcomes were the percent change in LDL-c, non-HDL-c, VLDL-c, lipoprotein-associated lipase A2 (Lp-PLA2), and apolipoprotein B from baseline to 12 weeks. Laboratory measurements were analyzed by a central laboratory.
In the REDUCE-IT trial, the primary efficacy outcome was the time from randomization to the first occurrence of any of the composite outcome — blindly adjudicated by an independent committee — that incorporated the following: CV death, non-fatal MI, non-fatal stroke, coronary revascularization, and hospitalization for unstable angina. Researchers assessed the time from randomization to the first occurrence of any component of the composite outcome. All observed data that were positively adjudicated by the committee, including data from patients with premature discontinuation of study drug, were included in the primary analysis. All components of the composite outcome were evaluated for up to 6.2 (median 4.9) years. The key secondary outcome was the time from randomization to the first occurrence of a composite of CV death, non-fatal MI (including silent MI), or non-fatal stroke. Other secondary outcomes were the time from randomization to the first occurrence of the individual or composite of CV death or non-fatal MI; fatal or non-fatal MI; non-elective coronary revascularization; CV death; unstable angina; fatal or non-fatal stroke; composite of total mortality, non-fatal MI, or non-fatal stroke; and total mortality. Morbidity events related to heart failure and cardiac arrhythmias were pre-specified as important outcomes for this review. Time from randomization to the first occurrence of these were tertiary outcomes in REDUCE-IT.
Adverse events were assessed in both studies using accepted definitions and approaches. No minimally important difference was set for any of the continuous outcomes in both studies.
Statistical Analysis
The primary efficacy variable in the ANCHOR trial was the percent change in fasting TG levels from baseline to week 12. A sample size of 194 patients per treatment group provided 90.6% or greater power to detect a difference of 15% between icosapent ethyl 4 g daily and placebo in percent change from baseline in fasting TG levels, assuming a standard deviation (SD) of 45% in TG measurements and a significance level of P < 0.05. To account for a 10% dropout rate, a total of 216 patients per treatment group were needed. For efficacy parameters, baseline (visit 4 [week 0]) measurement and week 12 (visit 7) differences were compared between groups. The primary efficacy (ITT) analysis was performed using an analysis of covariance (ANCOVA) model with treatment, sex, type of statin, and presence of diabetes as factors, and baseline TG value as a covariate. The Wilcoxon rank sum test was used as an alternative non-parametric analysis for the treatment comparisons, and medians and quartiles were provided for each treatment group. Estimates for the median of the treatment differences and 95% CI were provided for each treatment comparison. Authors used the Hommel’s procedure to test the adequate control for type I error for multiple comparisons for secondary end points.
In the REDUCE-IT trial, sample size was determined by estimating the adjudicated primary end point events. With 90% power to detect a 15% lower relative risk reduction of the primary composite end point in the icosapent ethyl group than in the placebo group, approximately 1,612 events would be required, and the sample size needed to reach this number of events was approximately 7,990 patients.
The REDUCE-IT study assessed the primary outcome by counts and Kaplan–Meier estimates of the percentage of patients experiencing each type of event by study completion per treatment arm. HRs and 95% CIs were generated with the use of a Cox proportional hazards model that included trial-group assignment as a covariate, stratified according to CV risk category, geographic region, and use of ezetimibe. The two-sided alpha level for the primary analysis was adjusted to 0.0437 from 0.05 to account for the two interim analyses based on a group sequential design with O’Brien–Fleming boundaries generated using the Lan-DeMets alpha-spending function. Log-rank P values from the Kaplan–Meier analysis (stratified based on the three randomization factors) are reported. Subgroup analysis was performed using Kaplan–Meier estimates and the log-rank test stratified by stratification factors used at randomization (except where the subgroup was a stratification factor). The subgroups of interest for this review were pre-specified subgroups in REDUCE-IT: baseline CV risk category (primary versus secondary prevention) and presence or absence of diabetes at baseline. Tests for interaction between subgroup categories were performed and considered a P value of less than 0.15 to be statistically significant. The sponsor noted that subgroup analyses were not powered to detect statistically significant differences between treatment groups within each individual subgroup, “particularly when a subgroup represents less than 50% of the enrolled population, and when a subgroup has low event rates.”
The key and other secondary outcomes and tertiary outcomes, as well as the components of the composite outcomes, were analyzed using the same methods as the primary outcome analysis. Statistical analyses of secondary outcomes followed a hierarchical sequential approach to control for inflated type I error. Specifically, the key secondary end point (the time from randomization to the first occurrence of the composite of CV death, non-fatal MI [including silent MI], or non-fatal stroke) was tested only if the primary analysis was statistically significant. Other secondary end points were the time from randomization to the first occurrence of the individual or composite end points, as follows (statistically tested in the order listed):
composite of CV death or non-fatal MI (including silent MI)
fatal or non-fatal MI (including silent MI)
non-elective coronary revascularization
CV death
unstable angina requiring emergent hospitalization
fatal or non-fatal stroke
composite of total mortality, non-fatal MI (including silent MI), or non-fatal stroke
total mortality.
Testing was done at a significance level of 0.0437 and ceased when a comparison for a secondary end point was greater than this threshold. All analyses beyond the primary or the last end point meeting statistical significance in this hierarchical order at this alpha level were exploratory, per the analysis plan.
Analysis Populations
For the ANCHOR study, the following analysis populations were defined:
The ITT population included all randomized patients who took at least one dose of any study drug, had a valid baseline laboratory efficacy measurement, and had at least one valid post-randomization laboratory efficacy measurement of any type. The ITT population was the primary analysis population for the efficacy analyses.
The per-protocol population included all ITT patients without any major protocol deviations. The per-protocol population was used to assess robustness of the primary analysis results.
The safety population included all randomized patients who received at least one dose of any study drug.
For the REDUCE-IT study, the following analysis populations were defined:
The ITT population was defined as all patients who were randomized. All efficacy analyses, including the primary analysis, were performed on the ITT population.
The modified ITT population was defined as all randomized patients who had study drug dispensed after randomization. Patients were analyzed according to the randomized treatment.
The per-protocol population included all modified ITT patients without any major protocol deviations who had 80% or greater adherence while on treatment. To be included in the per-protocol population, the minimum time on therapy was 90 days.
The safety population was defined as all randomized patients, and was the same as the ITT population. Patients were analyzed for safety according to treatment received.
Results
Patient Disposition
In the ANCHOR study, 2,309 patients were screened and 702 were eligible for randomization (30.4%), of which 233 patients were randomized to the icosapent ethyl 4 g daily group, 236 to icosapent ethyl 2 g daily group, and 233 to the control (placebo) group. No major differential dropouts were noticed during the analysis of the disposition of patients (5.2% versus 6.9%, respectively; ).
In the REDUCE-IT study, there was a large number of screening failures. From 19,212 screened patients, 11,033 (57.4%) were not included in the randomization schedule, mostly due to not meeting inclusion criteria, withdrawal of consent, adverse events before randomization, and loss to follow-up. Of the patients eligible for randomization, 4,089 were assigned to the icosapent ethyl group and 4,090 to the control group. Also, little to no difference in the number of patients who discontinued medications between groups was found (9.9% versus 11.2%, respectively, ).
Exposure to Study Treatments
In the ANCHOR study, drug adherence (exposure) or days of possible exposure was defined as the date of last dose of study drug: date of first dose + 1 during the double-blind treatment period. Numbers were also presented as percentages. Overall, 88.2% of patients reached more than 90% of exposure, with 88.2% in the icosapent ethyl group and 89.7% in the control group reaching this exposure (). Approximately 85% of patients were adherent to statin therapy, and 83% remained on the same statin dose throughout the study; there were no differences between treatment groups related to statin use.
Overall Study Treatment Exposure (ANCHOR Study).
In the REDUCE-IT study, drug exposure was calculated as the number of doses assumed to be taken relative to documented dosing period — from randomization to the patient’s final date in the study. Overall, 91.9% of patients in the icosapent ethyl group and 91.2% in the placebo group were at least 80% compliant with study drug (i.e., took at least 80% of their prescribed study drug capsules during the study). shows the treatment exposure for the REDUCE-IT study. Approximately 3% of patients in both treatment groups were not adherent with study statin use (i.e., took less than 80% of their prescribed statin during the study), and approximately 0.1% of patients in both groups were not on a stable statin regimen during the study. Less than 4% of patients in each treatment group used fibrates, niacin, bile acid sequestrants, PCSK9 inhibitors, or omega-3 fatty acid compounds after randomization during the study.
Overall Study Treatment Exposure (REDUCE-IT Study — Safety Population).
Efficacy Outcomes
All-Cause Mortality
Based on the REDUCE-IT study, icosapent ethyl did not statistically significantly reduce overall mortality. The event rates were 6.7% in the icosapent ethyl group versus 7.6% in the control group (HR 0.87; 95% CI, 0.74 to 1.02). The ANCHOR study did not evaluate this outcome.
Cardiovascular Mortality
Based on the REDUCE-IT study, a reduction in the rate of CV mortality (includes adjudicated CV deaths and deaths of undetermined causality) was observed, with a 4.3% event rate in the icosapent ethyl group versus 5.2% in the control group (HR 0.80; 95% CI, 0.65 to 0.98).
Non-Fatal Cardiovascular Events
Non-fatal CV events included non-fatal MI (including silent MI) and non-fatal stroke. The REDUCE-IT study showed that using icosapent ethyl results in a reduction in MI, with an event rate of 5.8% in the icosapent ethyl group versus 8.1% in the placebo group (HR 0.69; 95% CI, 0.59 to 0.83). Also based on this study, icosapent ethyl probably reduces the rate of non-fatal strokes (2.1% events in the intervention group versus 2.9% in the placebo group; HR 0.70; 95% CI, 0.53 to 0.93).
Hospitalizations Due to Unstable Angina, Heart Failure, and Arrhythmia
Icosapent ethyl reduced the occurrence of hospitalizations due to unstable angina (2.6% versus 3.8% event rate in the intervention versus placebo groups, respectively; HR 0.67; 95% CI, 0.53 to 0.86) in REDUCE-IT.
Icosapent ethyl was not found to be different from placebo for the risk in hospitalizations due to congestive heart failure (3.4% versus 3.5% event rate in the intervention and placebo groups, respectively; HR 0.97; 95% CI, 0.77 to 1.22) or cardiac arrhythmias (4.6% versus 3.8% event rate in the intervention and placebo groups, respectively; HR 1.21; 95% CI, 0.97 to 1.49).
Revascularization
Total coronary revascularizations were reduced with the use of icosapent ethyl (event rate of 9.2%) versus placebo (13.3%) (HR 0.66; 95% CI, 0.58 to 0.75). Total revascularization was not part of the hierarchical analysis plan and therefore not adjusted for inflated type I error.
Health-Related Quality of Life
Neither study evaluated the effects of icosapent ethyl on health-related quality of life.
Lipid Blood Levels and hsCRP
Both studies assessed blood concentrations of lipids and hsCRP. From the REDUCE-IT study, icosapent ethyl reduced TG levels, LDL-c, HDL-c, and hsCRP (). When comparing these changes between intervention and placebo groups, the icosapent ethyl group showed larger differences.
The ANCHOR study’s main efficacy outcome was the mean change from baseline in TG and other lipids. The icosapent ethyl group had a larger reduction in total TG levels from baseline values (measured at 12 weeks of follow-up) than the placebo group (21.5% median difference between groups). The same was observed with levels of LDL-c, HDL-c, and hsCRP; these changes from baseline were statistically significant ().
Efficacy Outcomes — Lipids and hsCRP (ANCHOR Trial).
Composite Outcomes
The primary and key secondary composite outcomes used in the REDUCE-IT study were not pre-specified outcomes in the protocol for this review. The outcomes, however, consist of individual outcomes that were identified as relevant, the results for which have already been presented.
shows the results for the primary and key secondary composite outcomes. Icosapent ethyl reduced the risk of both outcomes versus placebo. The Kaplan–Meier curve for the primary outcome is presented in . Sensitivity analyses confirmed the primary outcome results (Appendix 3, Table 16).
Mortality and Non-Fatal Events — ITT Population.
Kaplan–Meier Curve of Time to Primary Composite End Point From Date of Randomization (REDUCE-IT Trial). CI = confidence interval.
The occurrence of the primary outcome was lower with icosapent ethyl than with placebo in the established CVD (secondary prevention) subgroup, but there was no statistically significant difference between groups in the subgroup at risk for CVD (primary prevention) (Appendix 3, Table 16). The test for interaction between these subgroups was statistically significant (P = 0.1388). In the subgroup of patients with diabetes at baseline and in those without diabetes at baseline, icosapent ethyl reduced the occurrence of the composite primary outcome relative to placebo to a similar magnitude (P value for interaction = 0.5598).
Results from the analyses of the individual components of the primary and key secondary outcomes, each analyzed as an independent outcome (e.g., time to first occurrence of non-fatal MI, regardless of the time to first occurrence of any other end points for the same patient), is presented in (Appendix 3).
Harms
Adverse Events
The percentage of patients experiencing AEs was similar in the icosapent ethyl and placebo groups in REDUCE-IT (82% and 81%, respectively) and in ANCHOR (45% and 48%, respectively). The most common adverse events reported in both studies (with prevalence above 3%) were also similar between groups. Among these, the most commonly reported were diarrhea, nausea, back pain, nasopharyngitis, and arthralgia ( and ).
Summary of Harms — REDUCE-IT Study.
Summary of Harms — ANCHOR Study.
Serious Adverse Events
The REDUCE-IT study reported that 31% of patients in each group had a serious adverse event. In the ANCHOR trial, 3% of patients in the icosapent ethyl group and 2.1% in the placebo group had a serious adverse event. Individual types of serious adverse events occurred at a frequency of less than 3% in REDUCE-IT.
Withdrawals Due to Adverse Events
There was no difference between the icosapent ethyl and placebo groups (8% in each group) in the number of patients who withdrew due to adverse events in the REDUCE-IT study. Fewer patients in the ANCHOR study withdrew due to adverse events (2.1% with icosapent ethyl and 3.0% with placebo).
Notable Harms
Potential adverse events of particular interest were identified for the review based on feedback from clinical experts, the draft product monograph for icosapent ethyl, and notable harms pre-specified in the protocol. These notable harms included bleeding leading to transfusion or hospitalization (including visits to the emergency department), edema, atrial fibrillation, constipation, gout, musculoskeletal pain, arthralgia, and diarrhea ( and ).
In the REDUCE-IT study, a larger percentage of patients treated with icosapent ethyl had atrial fibrillation than in the placebo arm (5.3% versus 3.9%, respectively). Only one patient with atrial fibrillation was reported in the ANCHOR study, in a person randomized to placebo. Peripheral edema was also observed more frequently in the icosapent ethyl group than in the placebo group in REDUCE-IT (6.5% versus 5.0%, respectively) and in the ANCHOR study (1.3% versus 0.9%, respectively). Rates of serious adverse bleeding events were 2.7% in the icosapent ethyl group and 2.1% in the placebo group in REDUCE-IT; there were no fatal bleeding events in either group. There were no differences in the rates of adjudicated hemorrhagic stroke between the icosapent ethyl group and the placebo group.
A higher frequency of constipation was reported for icosapent ethyl versus placebo (5.4% versus 3.6%, respectively) in REDUCE-IT. Diarrhea was the most commonly occurring AE in both trials but occurred more frequently in the placebo arm than in the icosapent ethyl group. Musculoskeletal pain (back pain and arthralgia) occurred more frequently with icosapent ethyl than placebo in the trials. Finally, gout was observed in more patients treated with icosapent ethyl than placebo in REDUCE-IT; however, in ANCHOR, two patients in the placebo group had gout, whereas no patients treated with icosapent ethyl had gout.
Critical Appraisal
Internal Validity
Both studies had a proper randomization process. The generation of the randomization sequence was adequate, and the concealment of the allocation sequence was concealed until participants were enrolled and assigned to the interventions. Furthermore, no differences were noted in baseline characteristics, suggesting that the randomization process was successful.
The blinding of participants, clinicians, and researchers was achieved through identical placebo capsules, which avoided important and unbalanced deviations from the intended interventions. There is no clear evidence that participants were aware of their assigned intervention during the trial. Additionally, patients who discontinued or deviated from the interventions were properly analyzed in both ITT and per-protocol principles. Certain scenarios related to censoring in the primary time-to-event analysis were modelled appropriately in REDUCE-IT.
Overall, follow-up was relatively complete for the primary end point, with more than 95% of patients accounted for in the ITT analysis. Missing data were handled by evaluating both ITT and per-protocol analyses. Differences in missing data between study groups were unlikely to affect the final results.
The ANCHOR trial was short-term, and long-term effects could not be evaluated. However, outcomes were objectively obtained, and the processes to accomplish outcome measurements were well described. In the REDUCE-IT trial, the components of the composite outcome were objectively assessed in a blinded fashion by an appropriate adjudication committee to minimize bias. These measurements were similar among intervention and placebo groups.
There is a low risk of bias due to selection of the reported results. A protocol is well described for both studies, and the results analyzed are in accordance with the pre-specified analysis plan, even after considering the amendment in the protocol of the REDUCE-IT study to designate a key secondary composite outcome. This amendment was performed before the study’s conclusion and data evaluation.
Subgroup analyses were set a priori and properly conducted. However, all were underpowered to detect a significant effect from modifiers. Multiplicity was assessed by using a hierarchical model; however, it was not accounted for in the subgroup analysis.
The placebo used in the REDUCE-IT study was composed of mineral oil to mimic the active intervention. An increase in LDL-c levels at year one was observed in the placebo group when compared to the icosapent ethyl–treated arm (an increase of 10.2% and 3.1% in the placebo and intervention groups, respectively; ). Also, an increase in hsCRP at year two was reported (32.9% versus −13.9% in the placebo versus intervention groups, respectively) suggesting a possible biological effect of the mineral oil contained in the placebo, potentially inhibiting the gastrointestinal absorption of statins. These increases were not substantial, and a post hoc analysis by the sponsor suggests a similarly lower risk of events with icosapent ethyl and placebo, regardless of whether there was an increase in LDL-c in the placebo arm. However, the clinical experts consulted by CADTH indicated that this was notable.
Efficacy Outcomes — Lipids and hsCRP (REDUCE-IT Trial), ITT Population.
External Validity
Patients included in the ANCHOR study were only from the US, while those from the REDUCE-IT trial were distributed worldwide. Results from the primary outcome by region subgroup analysis (categorized as a group of western countries, Eastern European countries, and Asia–Pacific) in REDUCE-IT suggested potential differences in the magnitude of treatment effects by region (HR 0.491 for Asia–Pacific region, 0.740 for western countries, and 0.842 for Eastern European countries); however, the numbers of patients in the Eastern European and Asia–Pacific subgroups were relatively small, with imprecise estimates (very wide confidence intervals), and the test for interaction was not statistically significant (P = 0.3046). The clinical experts consulted by CADTH indicated that, despite some potential differences, the populations are likely similar to the target population in Canada in which icosapent ethyl would be used.
As in many clinical trials, the patients included could be considered highly selected, as the studies excluded a large number of patients (screening failures) who could be encountered in real clinical practice, such as those with TG below 2.3 mmol/L (200 mg/dL) or above 5.6 mmol/L (500 mg/dL), with congestive heart failure, active liver disease, or a planned coronary surgery or intervention. The mean (SD) ages in both studies were approximately 61 (9.8) and 63 (8.4) years; the clinical experts noted that the studies provide limited evidence on patients younger than 50 years and older than 70 years, which are patients seen in clinical settings.
The distribution of the baseline statin intensity may not reflect clinical practice. The clinical experts consulted by CADTH indicated that the distribution of patients receiving moderate- and high-intensity statin therapy is reversed from what clinical practice guidelines would recommend in this population of patients. The goal should be to first ensure optimal treatment with a statin before adding another therapy: the highest tolerated intensity of statin therapy is used to bring the lipid profile to the target range. The experts acknowledged that the distribution in the studies may reflect what is observed in registry data, because there is suboptimal adherence to practice guidelines in clinical practice settings. The distribution of baseline statin intensity was balanced between treatment groups in both studies and was unlikely to influence the results; furthermore, regardless of statin intensity distribution, the median baseline LDL-c level was within target levels at approximately 1.94 mmol/L (75 mg/dL) across the studied population.
Other Relevant Studies
At the time of writing this report, no other relevant studies have been published from the REDUCE-IT study. Regarding the ANCHOR study, one report pertinent to this review is described below.
Ballantyne et al.28 is an exploratory analysis pre-specified from the ANCHOR study. It assesses patients in the same group with TG levels between 2.3 and 5.6 mmol/L (200 and 500 mg/dL) despite statin treatment. However, the aim of the study was to assess the effects of icosapent ethyl on lipoprotein particle concentration and size as the main outcome, and the correlations of atherogenic particles with apolipoprotein B. Nuclear magnetic resonance spectroscopy was used to measure lipoprotein particle concentration and size. It was reported that, compared with placebo, icosapent ethyl 4 g per day significantly reduced VLDL-c (7.7%, P = 0.0001) and HDL-c (1.2%, P = 0.0014) particle sizes, with a modest but significant increase in LDL-c particle size (0.5%, P 0.0031). This is a descriptive study with indirect outcomes that are not directly applicable to clinical practice.