Internal Validity
VISIBLE 2 was the only pivotal study included in this review. The investigators adequately produced a randomization sequence, with a proper concealment of the random sequence using a central randomization scheme under the supervision of the sponsor, by means of an IWRS until participants were enrolled and assigned to the interventions. Differences were noted in the baseline characteristics of patients, such as prior anti–TNF alpha use, concomitant medications with immunomodulators only at week 0 and ileal disease. These differences were small and less likely to have a meaningful impact on the validity of the results; however, the higher proportion of patients with prior anti–TNF alpha use and ileal disease only, and the fewer patients who received concomitant immunomodulators only at baseline in the vedolizumab SC group compared to placebo may suggest that patients in the vedolizumab group would be more difficult to treat, which could result in a more conservative estimate of the treatment effect. The blinding of participants, clinicians, and researchers was achieved through identical placebo and vedolizumab presentations, which avoided important and unbalanced deviations from the intended interventions. There is no evidence that participants were aware of their assigned intervention on the double-dummy design of the trial. Patients who stopped or deviated from the interventions were properly accounted for and analyzed in an FAS, which was close to the ITT population in the study.
Multiplicity was properly considered, and adequate tests were conducted (i.e., a hierarchical approach was used) to control for an overall type I error rate.
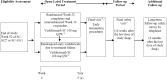
During the maintenance phase, 41% of the participants prematurely discontinued the study drug, 45% in the placebo group and 39% in the vedolizumab SC group. The main reason for treatment discontinuation in the maintenance phase was lack of efficacy (with 71% and 73% on placebo and vedolizumab SC among those who discontinued, respectively), followed by voluntary withdrawal and AEs. This difference in missing data could bias the results. Sensitivity analyses were conducted to examine the robustness of study findings to missing data assumptions, ▬▬▬▬▬.
Outcomes were objectively obtained with validated tools (see Appendix 1) and the processes to carry out outcome measurements were well described and assessed in a blinded fashion. There is a low risk of bias due to selection of the reported results. A protocol was well described, and the presented results follow the pre-specified analysis plan. Amendments made during the study were well addressed and unlikely to affect the end results or imply bias due to selection of participants.
Subgroup analyses were performed to examine the consistency of the treatment effect observed for the primary and all secondary outcomes based on age, gender, race, duration of CD, geographic region, baseline disease activity, baseline fecal calprotectin, disease localization, clinical remission status at week 6, prior TNF alpha antagonist therapy, prior immunomodulator and TNF alpha antagonist failure, prior corticosteroid failure, prior immunomodulator failure, concomitant therapies, and worst prior treatment failure. However, conclusions in regard to these subgroups are uncertain due to the small sample sizes in the subgroups. In addition, subgroup analyses were exploratory in VISIBLE 2, and there was also a lack of adjustment for multiplicity. All of these increase the uncertainty in interpreting the results in the subgroups. Appendix 2 presents the efficacy outcomes by prior TNF alpha antagonist therapy (patients without prior exposure to TNF alpha antagonist therapy versus patients who had prior exposure to TNF alpha antagonist therapy but did not fail this treatment versus patients who had prior failure to TNF alpha antagonist therapy).
The VISIBLE 2 study was powered to assess the primary outcome of clinical remission after 52 weeks but was not sufficient to assess other secondary end points. This limitation contributed to the findings of numerically greater but not statistically significant differences between treatment arms for all secondary end points, such as enhanced clinical response and corticosteroid-free clinical remission.
External Validity
The populations included in VISIBLE 2 are, to an extent and within the limitations of a controlled setting of a clinical trial, similar to what it is encountered in clinical practice and relevant to the population of interest for this review, which focuses on SC administration and specific doses that are in accordance with what is approved by Health Canada and planned to be used in real-life practice. However, adherence could be overstated as it is usual in controlled randomized trials, and generalizability may be an issue when the medication is applied in real clinical settings.
The amount and type of co-interventions allowed during the study can be considered close to what happens in clinical practice, although more frequent clinical visits and assessments can be overestimated. Patients needed training to apply the SC vedolizumab doses and the study participants reportedly performed well in this sense. It is likely this training would be similar to real clinical practice.