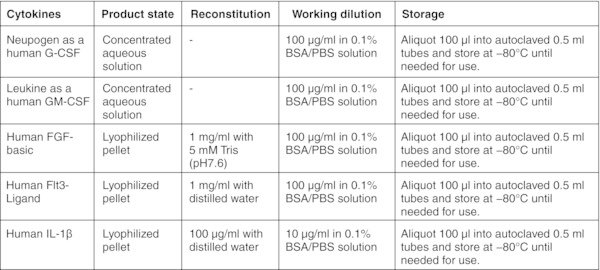
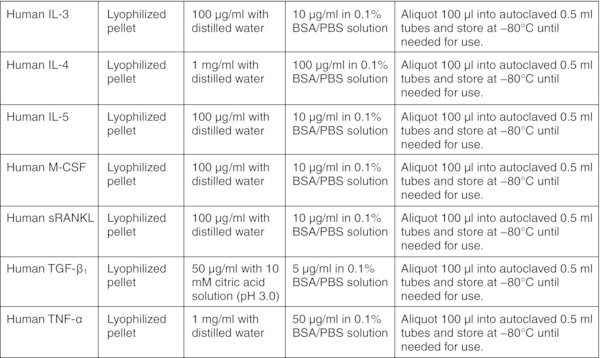
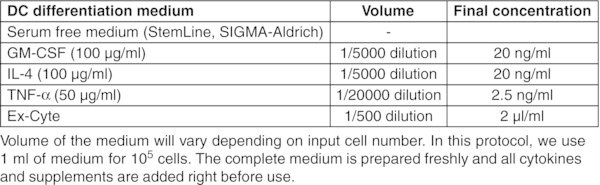
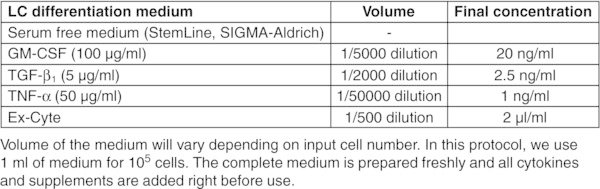
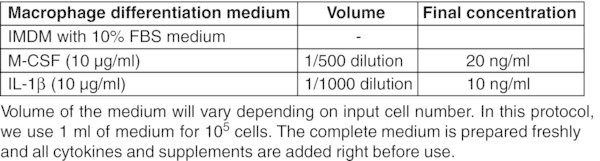
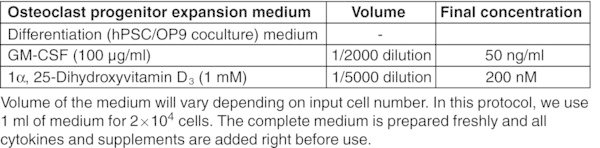
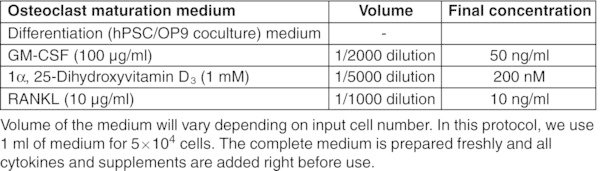
CRITICAL: Perform entire procedure in a sterile biosafety cabinet. Read the product information sheet carefully before preparation of working aliquots of all cytokines.
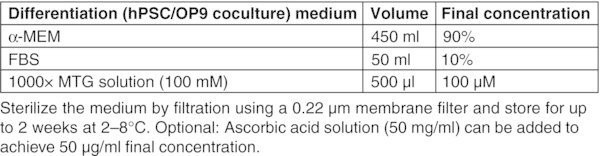
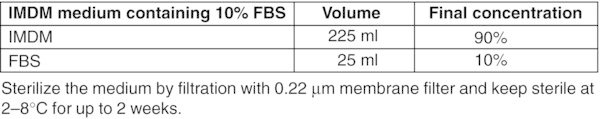
All basal medium, α-MEM, DMEM, DMEM/F-12, and IMDM should be prepared freshly from powder according to the manufacturer's instructions, sterilized by filtration using a 0.22 μm membrane filter and stored for up to 2 months at 2–8°C. Figures:
Inactivate MEFs with gamma irradiation at 8,000 rad
Resuspend MEFs at 2×105 cells/ml in prewarmed MEF growth.
Add 2 mL/well of prewarmed MEF growth medium and then dispense MEF suspension on gelatin- coated 6-well plate (1 ml/well). CRITICAL STEP Distribute MEFs evenly with a back/forth and right/left movement twice. Irregular distribution of MEFs may cause death and unwanted differentiation of hPSC colonies during culture.
Incubate MEF plates in a CO2 incubator at 37°C for at least 24 hrs before adding hPSCs. CRITICAL STEP MEFs should be used for hPSC passage within one week. Before plating hPSCs, aspirate MEF medium, add 2 ml of PBS, swirl once and aspirate PBS. Add 2 ml of prewarmed hPSC medium and place plate into CO2 incubator at 37°C. Now MEF feeders are ready for hPSC plating (step 15).
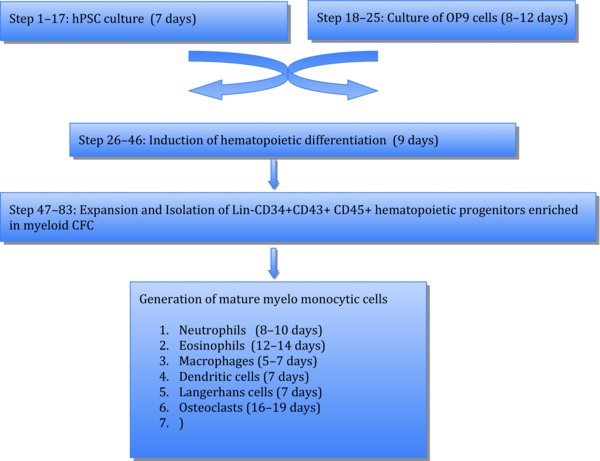
hES/iPSC culture TIMING 7 days
Aspirate hPSC growth medium from one well of the 6-well plate of hESCs or iPSCs. CRITICAL STEP: Note that cells will need to be split every 6–7 days.
Wash cells with 2 ml/well of PBS (Ca2+ and Mg2+ free) stored at room temperature.
Add 1 ml/well of collagenase IV solution (1 mg/ml) and incubate at 37°C in a CO2 incubator until the edges of the hPSC colonies begin to curl (approximately 7–10 minutes).
Add 1 ml of hESC or hiPSC growth medium and break up the colonies into small cell aggregates by gently pipetting. CRITICAL STEP: After collagenase treatment, hPSC colonies are loosely attached and can be collected by gentle pipetting. Do not use excessive mechanical force or scraping which can provoke spontaneous differentiation.
Transfer cells to a 15 ml conical tube.
Centrifuge at 200×g at room temperature for 3–5 min.
Aspirate the medium gently without disturbing the pellet.
Resuspend cells in 3 ml of hESC or hiPSC growth medium, and wash cells by repeating steps 11 and 12.
Resuspend cell pellet in 3 ml of hESC or hiPSC growth medium.
Plate 0.5 ml/well of cell suspension onto MEF-grown 6-well plate from Step 5.
Troubleshooting
Feed hPSCs daily by replacing the old medium with 3 ml of prewarmed hESC or hiPSC medium.
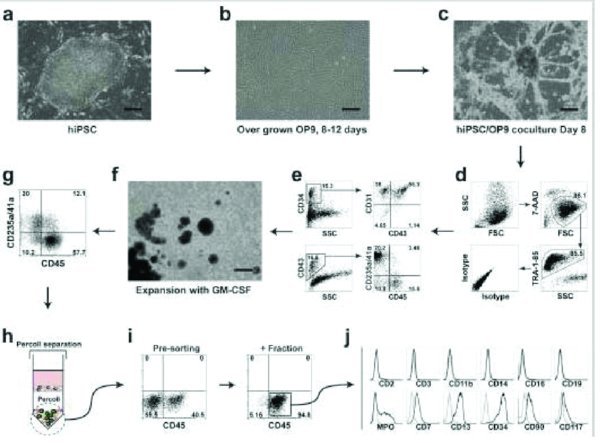
Passage undifferentiated hES/iPSCs (Fig. 1a) weekly at 1.2–1.5×10
6 cells/well density on MEFs. CRITICAL STEP: If spontaneous differentiation of hESCs or hiPSCs occurs, differentiated hPSC colonies should be eliminated during the maintenance; observe hPSCs every day before changing medium. Mark differentiated colonies with an objective marker under the inverted microscope and aspirate marked areas using a glass Pasteur pipette while feeding hPSC with fresh medium. CRITICAL STEP: Alternatively, hPSCs can be maintained under feeder-free conditions.
32 In OP9 coculture system, we did not observe significant differences in the efficiency of hematopoietic differentiation of hPSCs maintained on MEFs or in feeder-free cultures.
Culture of mouse OP9 cells TIMING 8–12 days
Aspirate OP9 growth medium and wash cells twice with 10 ml of PBS. CRITICAL STEP: Note that cells will need to be split every 4 days.
Add 5 ml of trypsin/EDTA(0.05%/0.5 mM) solution and incubate for 5 min at 37°C in a CO2 incubator. CRITICAL STEP: OP9 feeders consist of heterogeneous cell populations which include cells with at least adipogenic and osteogenic potential. To maintain a proper balance of cells following passage, OP9 feeders should be digested and detached completely by trypsin treatment. Inadequate washing of cells with PBS or using an old trypsin may results in partial detachment and enrichment in adipogenic cells.
Add 5 ml of OP9 growth medium and collect cells by pipetting.
Transfer cell suspension into a 15 ml conical tube and centrifuge for 5 min at 300×g at room temperature.
Aspirate supernatant and resuspend cells in 1 ml of OP9 growth medium.
Add 100 μl of cell suspension to 10 ml of OP9 growth medium and plate cells onto 10 cm gelatin-coated culture dishes. CRITICAL STEP: It is essential to culture OP9 on gelatin-coated plates to prevent spontaneous adipogenesis.
Troubleshooting
When cultures are confluent, split 1 dish for maintenance. This should occur after approximately 4 days of growth.
To prepare overgrown OP9 for coculture with hPSCs, change half of the medium after 4 days of culture on gelatin-coated plates and incubate for an additional 4–8 days to achieve a dense OP9 monolayer. CRITICAL STEP: OP9 should be split every 4 days for maintenance/expansion. OP9 used for hPSC differentiation should be fed with fresh media at confluence (day 4) and incubated for an additional 4–8 days to form a dense monolayer embedded in extracellular matrix.
Hematopoietic differentiation on OP9 TIMING 9 days
Remove overgrown OP9 dishes prepared for coculture from the CO2 incubator.
Aspirate OP9 growth medium.
Add 10 ml of differentiation medium and keep at 37°C in a CO2 incubator.
From one well of a 6-well hPSC plate from Step 17, aspirate hES/hiPSC growth medium. Add 1 ml of collagenase IV solution (1 mg/ml), and incubate cells for 10 min at 37°C.
Add 1 ml/well of differentiation medium directly to the well and break up colonies into small cell aggregates by gentle pipetting. Transfer cells into a 15 ml conical tube. CRITICAL STEP: hPSCs for differentiation studies should be prepared as small aggregates. Single hPSCs will not survive on OP9.
Centrifuge cells at 200×g for 3–5 min at room temperature.
Aspirate the medium gently without disturbing the pellet.
Resuspend the cell pellet with 1 mL of differentiation medium.
Add 1 mL of hES/hiPSC suspension to 1 OP9 dish prepared in steps 26–28. CRITICAL STEP: Efficiency of hematopoietic differentiation is significantly affected by the density of hPSCs plated on OP9. In our experience, the optimal plating density of hPSCs is 1.0–1.5 × 106 cells per 10 cm dish of OP9. To estimate the number of cells in a suspension consisting of small hPSC aggregates, one well of a 6-well plate can be used to prepare a single cell suspension by treatment with trypsin for cell counting. Alternatively, aliquots of hPSC aggregates can be collected, treated with trypsin, and counted.
Distribute cells evenly with a back/forth and right/left movement twice.
The following day (day 1), aspirate all of the media to waste and replace with 20 ml of prewarmed differentiation medium.
On day 4, change half of the medium.
On day 6, change half of the medium
To collect cells on day 9, aspirate the supernatant and add 5 ml of prewarmed collagenase solution (1 mg/ml) to each dish of hPSC/OP9 coculture and incubate for 30 minutes at 37°C in a CO2 incubator.
Remove the collagenase solution and keep it on ice in a 15 ml conical tube for subsequent collection of trypsin digested cells (cell collection tube). CRITICAL STEP: hPSC/OP9 coculture produces collagen-rich matrix and pretreatment with collagenase is essential to achieve efficient digestion of cells with trypsin. Because some cultures might form an excessive amount of extracellular matrix, the time of treatment with collagenase can be extended up to 40–50 minutes to achieve complete dissociation and maximize cell recovery.
Add 5 ml of prewarmed Trypsin/EDTA solution (0.05%/0.5 mM) to the dish from Step 39 and incubate for 15–20 minutes at 37°C CO2 incubator.
Add 2 ml/dish of MACS buffer, suspend coculture cells by pipetting and transfer to the collection tube from Step 40.
Add an additional 5 ml/dish of MACS buffer to the coculture dish and collect the remaining cells into the collection tube.
Centrifuge cell suspension at 300×g for 5 min at room temperature.
Wash cells once by adding 5 ml of MACS buffer to cell pellet followed by pipetting and centrifugation at 300×g for 5 min at room temperature.
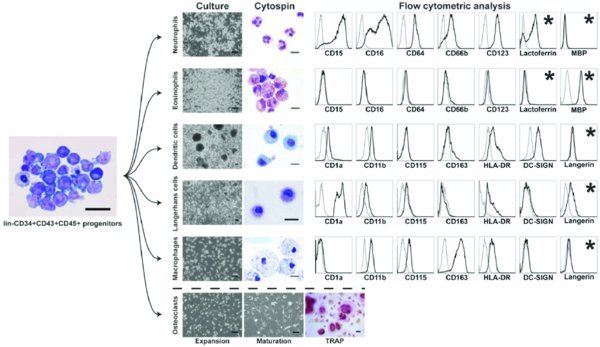
Cells are ready to use in further applications such as flow cytometry, CFC assay, and further differentiation. CRITICAL STEP: The success of subsequent steps in this differentiation protocol largely depends on effective induction of hematopoietic differentiation and lin-CD34+CD43+CD45+ progenitors in coculture with OP9. Therefore analysis of CD43 expression and simultaneous detection of CD235a/CD41a+ and CD45+ cells within CD43+ population can be performed to confirm myeloid commitment28 (see Fig. 1). CRITICAL STEP: Because we observed a decline in the hematopoietic differentiation capacity of hESCs after passage 50, we do not recommend the use of hESC lines beyond this passage. OP9 cells should not be used beyond passage 60, because of significant decrease in hematopoiesis-inductive potential.
Troubleshooting
Short-term expansion of multipotent myeloid progenitors TIMING 2 days
Wash out pHEMA-coated flasks with 20 ml (T75 flask) PBS.
Resuspend differentiated hPSCs (from Step 46) in multipotent myeloid progenitor expansion medium at a concentration of ∼1×106 cells/ml. CRITICAL STEP: Note that typically, 1.5–2×107 cells are recovered from one 10 cm dish of hPSC/OP9 coculture. We usually culture cells collected from 2 dishes in one T75 flask. GM-CSF is a single key factor required for expansion of hPSC-derived myeloid progenitors. The addition of SCF and/or FLT3L to expansion cultures has little effect on the growth of myeloid precursors, but significantly increases the proportion of CD235a+ erythroid cells.
Incubate 2 days at 37°C in a CO2 incubator. CRITICAL STEP: Differentiated hPSCs cultured in non-adherent conditions spontaneously reaggregate and form large floating cellular conglomerates with myeloid progenitors proliferating in suspension as single cells.
Purification of multipotent myeloid progenitors TIMING 3 hrs
Collect myeloid cultures (from Step 49) and filter through a 70 μm cell strainer into a 50 ml tube to remove cell aggregates.
Pellet the cells by centrifugation at 250×g for 5 min at room temperature
Resuspend cells with 5 ml of MACS buffer in a 15 ml tube.
Underlay cell suspension with 1.0–1.5 ml of Percoll solution. Place a 1 ml plastic serological pipet filled with Percoll solution into the tube with cell suspension, so the pipet tip touches the bottom of the tube. Very carefully dispense Percoll solution to underlay cell suspension avoiding mixing.
Centrifuge tube at 300×g for 10–15 min at room temperature.
Aspirate supernatant and interface containing dead cells and debris.
Resuspend cells with 5 ml of MACS buffer and centrifuge at 250×g for 5 min at room temperature. Take an aliquot of the resuspended cells before centrifugation to count the number of isolated cells.
Aspirate supernatant and add 0.2 ml of MACS buffer to cell pellet.
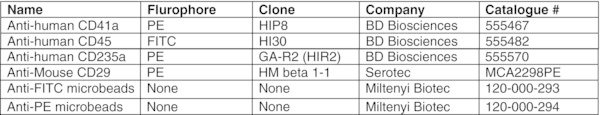
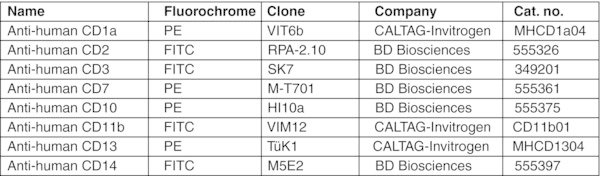
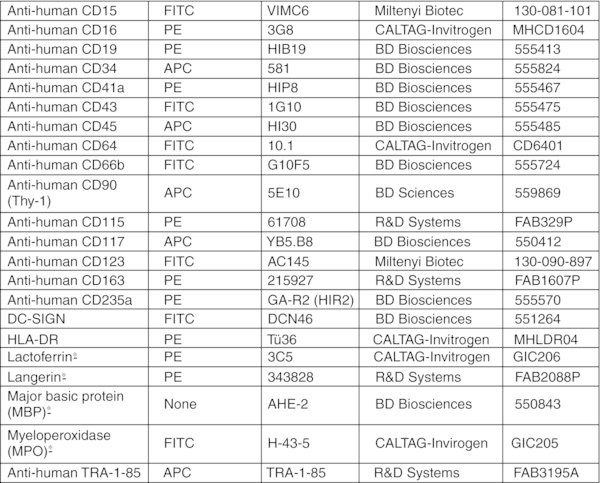
Add 1 μl of anti-human CD235a-PE, and 5 μl of anti-human CD41a-PE antibodies per 106 cells. CRITICAL STEP: Usually cells collected after Percoll separation are free of residual OP9 cells. If significant contamination of human cells with OP9 cells occurs, 10 μl of anti-mouse CD29-PE Ab can optionally be added to the cell pellet to deplete the mouse cells. The presence of contaminating OP9 cells in suspension can be evaluated by flow cytometry using mouse-specific CD29 antibodies.28
Set up tube on the MACS mixer and incubate at the lowest rotation speed at 4°C for 15–20 minutes.
Wash cells with ice-cold MACS buffer by adding 5 ml of MACS buffer to cell pellet followed by pipetting and centrifugation at 300×g for 5 min at 4°C, resuspend in 0.4 ml of MACS buffer, and add 10 μl of anti-PE magnetic beads.
Repeat step 59.
Wash cells with ice-cold MACS buffer as described in step 60 and resuspend in 1 ml of MACS buffer.
Filter cells through a 30 μm pre-separation filter. CRITICAL STEP: It is important to filter cells before magnetic separation to remove large cell aggregates which may block the magnetic column.
Assemble the MACS-LD separation column according to the manufacturer's instructions.
Wash column with 2 ml of MACS buffer.
Apply the cell suspension from Step 63 to the LD column allowing cells to pass completely through the column into 15 ml collection tube.
Wash column with 2 ml of MACS buffer and collect in same collection tube.
Recap and remove collection tube with unlabeled cells (CD235a-CD41a- human cells).
Centrifuge cells at 300×g for 5 min at 4°C.
Aspirate supernatant and add 80 μl of MACS buffer and 20 μl of anti-human CD45-FITC Ab.
Place the tube on the MACS mixer and incubate at the lowest rotation speed at 4°C for 15–20 minutes.
Wash cells with ice-cold MACS buffer as described in step 60, resuspend in 80 μl of MACS buffer, and add 20 μl of anti-FITC magnetic beads.
Repeat step 69.
Wash cells with ice-cold MACS buffer as described in step 60 and resuspend in 1 ml of MACS buffer.
Filter cells through a 30 μm pre-separation filter.
Assemble MACS-LS separation unit according to the manufacturer's instructions.
Rinse column with 2 ml of MACS buffer.
To purify CD235a/CD41a−CD45+ multipotent myeloid progenitors, apply the cell suspension from Step 75 to the LS column allowing cells to pass completely through the column into collection tube.
Wash column with 2 ml of MACS buffer and collect in same collection tube then discard.
Remove the column from the magnet and place in an empty 15 ml tube.
Wash out CD45+ cells with 5 ml MACS buffer using the plunger supplied with column.
Centrifuge cells at 300×g for 5 min at 4°C.
Resuspend cells in 0.2 ml of MACS buffer and keep on ice. Cells are ready to use for further differentiation.
Troubleshooting
Differentiation of hPSC-derived myelomonocytic cells
Differentiate hPSC-derived lin−CD34+CD43+CD45+ progenitors using option A for neutrophils, option B for eosinophils, option C for macrophages, option D for DCs, option E for LCs, and option F for osteoclasts. Protocol for cytospin preparation and staining of differentiated cells is provided as option G.