Methods
Primary objectives of Trial of Oral Mandibular Advancement Devices for Obstructive sleep apnoea–hypopnoea
The primary objective was to determine whether or not MADs are more effective than no treatment and whether or not the level of MAD sophistication (bespoke, semi-bespoke and over the counter) influences outcomes for patients with mild to moderate OSAH.
Secondary objectives of the Trial of Oral Mandibular Advancement Devices for Obstructive sleep apnoea–hypopnoea
The secondary objectives were to produce a trial-based cost-effectiveness analysis to determine, from a NHS perspective, whether or not MADs are cost-effective compared with no treatment in mild to moderate OSAH, and whether or not the degree of MAD sophistication influences cost-effectiveness. It was also intended that the results would contribute to a comprehensive long-term cost–utility analysis (see Chapter 4).
Study design
The study was an open-label, four-treatment, four-period, randomised crossover trial comparing the clinical effectiveness and cost-effectiveness of three types of MAD {bespoke MAD (bMAD; NHS Oral-Maxillofacial Laboratory, Addenbrooke’s Hospital, Cambridge, UK), semi-bespoke [SleepPro 2™ (SP2); Meditas Ltd, Winchester, UK] and over the counter [SleepPro 1™ (SP1); Meditas Ltd, Winchester, UK]} and a no-treatment control for patients with mild to moderate OSAH (AHI of 5 events/hour to < 30 events/hour). Each 6-week period (4 weeks for no-treatment arm) comprised a 2-week acclimatisation phase, followed by a 4-week treatment phase. A 1-week washout period followed active treatments.
The study was reviewed and approved by the National Research Ethics Service Research Ethics Committee East of England – Cambridge Central (reference 10/H0308/4) and local (research consortia and primary care trust), ethical and research governance committees, and was registered as an International Standard Randomised Controlled Trial, number (ISRCTN) 02309506. The trial protocol can be accessed at www.thelancet.com/protocol-reviews/10PRT-4998.
Public and patient involvement
There was involvement from a patient in the study design and conduct, with input into production of patient information and other trial documentation, and membership of both the trial management group and the trial steering group. Although patient involvement in the Data Monitoring and Ethics Committee (DMEC) was arranged, the patient representative was not able to contribute to the meetings.
Participants
All newly referred or existing patients attending the Respiratory Support and Sleep Centre (RSSC), a tertiary care, specialist sleep centre, at Papworth Hospital (Cambridge, UK), were invited to be screened for eligibility in the trial if they were ≥ 18 years of age and had, or were suspected of having, mild to moderate OSAH (AHI 5 events/hour to < 30 events/hour), confirmed by either respiratory PSG (rPSG) (Embletta™; Embla Systems, Kanata, ON, Canada) or complete PSG, and who had symptomatic daytime sleepiness defined by an ESS score of ≥ 9. Potential patients did not require CPAP, as defined in NICE Technology Appraisal number 139,37 or they had refused CPAP or chose inclusion in TOMADO instead. Patients were excluded if they were pregnant or had any of the following:
central sleep apnoea as the predominant form of sleep-disordered breathing
coexistent sleep disorder, poor sleep hygiene or drug treatment considered likely to have a significant impact on symptoms (especially sleepiness) or assessment of MAD effectiveness
severe and/or unstable CVD judged by clinician to warrant immediate CPAP
other medical or psychiatric disorders judged likely to adversely interact with MADs or confound interpretation of its effectiveness
significant periodontal disease or tooth decay; partial or complete edentulism; presence of fixed orthodontic devices
temporomandibular joint pain or disease
clinical history suggestive of severe bruxism
restriction in mouth opening or advancement of mandible
respiratory failure
inability to give informed consent or comply with the protocol
previous exposure to MAD treatment
disabling sleepiness leading to significant patient-specific safety concerns.
Screening/baseline visit
Following signed consent and enrolment, a medical history and clinical examination were undertaken to establish eligibility. The clinical examination included height, weight, neck circumference, waist-to-hip ratio and BP. Patients completed the generic HRQoL questionnaire, medical outcomes Short Form questionnaire-36 items (SF-36),53 the disease-specific Calgary Sleep Apnoea Quality of Life Index (SAQLI)54 and the European Quality of Life-5 dimensions three-level version (EQ-5D-3L) for use as a utility measurement.55 In addition, they completed a Functional Outcome of Sleep Questionnaire (FOSQ)56 and the ESS questionnaire.15 All patients who satisfied the other inclusion/exclusion criteria underwent confirmatory rPSG, unless they had already undergone rPSG or inpatient PSG within the previous 4 weeks for clinical reasons. In that case, the clinical PSG output was used as a baseline value.
Interventions
Three different non-adjustable MADs representing currently available devices along a spectrum of complexity and cost were studied:
SleepPro 1™: a thermoplastic ‘boil and bite’ device fitted by the patient following the manufacturer’s printed instructions. The patient softened the device in hot water, placed it into his or her mouth and, having bitten down on it, advanced the mandible to an individually determined ‘comfortable’ position. The device was then manually moulded against the teeth and set by subsequent immersion in cold water. Rewarming allowed remoulding.
SleepPro 2™: a semi-bespoke device, formed from a dental impression mould used by the patient. At the screening/baseline visit patients were given an impression kit to mould at home and then send to the manufacturer in order for the SP2 to be made. The impression kit consisted of a SP1 with holes to allow the injection of dental putty. The patient was instructed to mould the SP1 (as for the SP1 device), then wear it for two nights to ensure optimum position and fit, remoulding if necessary. The patient then made up the putty and injected it into the SP1, before sending the resulting impression back to the manufacturer. The SP2 was produced from this mould. It was designed to grip the entire dentition. Thinner walls than the SP1 were intended to result in a more comfortable fit. Involvement of the patient’s dentist in taking the impression was suggested, but not considered to be essential or key to achieving the best fit by the manufacturer.
Bespoke device: a custom-made MAD professionally fitted by a specialist NHS oral–maxillofacial laboratory at Addenbrooke’s Hospital, Cambridge, UK. A positional ‘wax bite’ was taken from the patient and the degree of mandibular advancement (50–70% of the maximal protrusive distance from centric occlusion, i.e. the ‘normal’ bite where the teeth all interdigitate maximally) was determined. Upper and lower full dental impressions were taken in alginate by a suitably qualified dental professional and cast in dental stone. The casts were trimmed and articulated using the positional wax bite. A blow-down splint in soft acrylic was created on each cast and then fused with a further acrylic blow-down to ensure the upper and lower dentition were positioned in the predetermined optimal position to hold the mandible forward. The patient returned roughly 2 weeks later for the fitting. The fitting allowed for optimal balance between advancing the mandible sufficiently to bring the tongue base off the posterior pharyngeal wall and patient comfort.
Degree of protrusion
As this was a pragmatic trial, the SP1 and SP2 devices were both advanced by the patient, according to manufacturer’s instructions. The bMAD was fitted by qualified dental experts, who determined the degree of protrusion with the patient, aiming for maximal comfortable advancement. The aim was to advance the mandible by a minimum of 50% of maximal protrusion. The degree of protrusion of each device was measured by the trial team, where possible, at the end of the patient’s involvement in the study.
Patients started the first treatment arm following the manufacture of all of the MADs. The first 2 weeks of each treatment period were an acclimatisation phase to allow patients to adjust to each device and not considered part of active treatment. After 2 weeks, patients were telephoned to assess initial tolerability and adherence, and to record any contact with the research team, maxillofacial laboratory or other clinical staff in the previous 2 weeks. All patients received 4 weeks of treatment with each MAD and the no-treatment control, with outcome assessment at the end of each treatment period.
A 1-week no-treatment washout period followed each active treatment to avoid carryover effects. All MADs were kept at Papworth outside the treatment period and patients were asked to return each device at the end of the treatment assessment, and before starting the next treatment.
Outcome measures
Primary outcome measure
The primary outcome measure was the AHI, defined as the number of apnoea or hypopnoea events per hour of sleep. It was assessed by home rPSG using Embletta™ equipment following each treatment period. Airflow was measured using both a nasal air pressure transducer and an oronasal thermal sensor. All rPSG studies were scored manually in anonymised batches by a NHS polysomnographer, blinded to treatment allocation, in accordance with the AASM guidelines.12 Throughout the trial, 16% of sleep studies were scored in parallel by a second polysomnographer to ensure inter-rater agreement and adherence to recommended guidelines.
Secondary outcome measures
Subjective sleepiness (ESS): daytime sleepiness is a key feature of OSAH, resulting from disrupted sleep, and its effective control is a major aim of treatment. Patients are required to assess, on a 4-point scale (0, 1, 2 and 3), the likelihood of falling asleep during eight different daily activities (see
Appendix 1). Item scores are summed giving a range for the overall score of 0–24, with 0–9 classified as normal daytime sleepiness, 10–15 as mild daytime sleepiness and 16–24 as moderate/severe daytime sleepiness.
Physiological indices from rPSG: 4% ODI, mean, minimum and time spent < 90% of nocturnal oxygen saturation (SpO2).
Systolic BP (SBP) and diastolic BP (DBP).
Functional status (FOSQ): the FOSQ is a condition-specific functional status measure designed to evaluate the impact of disorders of excessive sleepiness on activities of daily living (see
Appendix 2). In total, there are 30 questions and five subscales: general productivity, social outcome, activity level, vigilance and intimate relationships and sexual activity. The total score can range from 5 to 20, with a lower score representing greater dysfunction. The potential range of scores for each subscale is 1–4, with a lower score representing greater dysfunction. The FOSQ was administered at baseline and after each of the four treatment periods.
Disease-specific HRQoL (SAQLI): the SAQLI is a condition-specific questionnaire to assess obstructive sleep apnoea-related QoL (see
Appendix 3). There are 14 questions and four domains. The total score can range from 1 to 7, with a lower score representing greater dysfunction. The potential range of scores for each subscale is also 1–7, with a lower score representing greater dysfunction.
Generic HRQoL using both the SF-36 and the EQ-5D-3L: the SF-36 has eight dimensions of HRQoL on a scale of 0 (minimum function) to 100 (maximum function), named: physical functioning; role limitations because of physical problems; pain; energy/vitality; social functioning; mental health; role limitations because of emotional problems; and general health (see
Appendix 4). These scales can be combined into two composite scales named the physical component score (PCS) and the mental component score (MCS).
57 We have adopted the commonly used standardisation method so that for a general population the PCS and MCS have mean 50 and SD 10. The EQ-5D-3L (see
Appendix 5) has five dimensions (morbidity, self-care, usual activities, pain or discomfort and anxiety or depression), each with three levels (no problems, a moderate problem or a severe problem).
Treatment adherence, hours of use and device retention as well as patient sleep duration (assessed by a daily sleep diary).
Snoring scale: partner-rated visual analogue scale (VAS).
Driving and RTA questionnaire (for economic modelling).
Side effects, withdrawals, patient satisfaction and device preference at trial exit.
Resource use: data on individual health-care resource use were collected on a study-specific case report form (see
Appendix 6). This included type of device, number of home/surgery visits [general practitioner(GPs), nurses] number of visits [dentists, accident and emergency (A&E), outpatients, additional visits to Addenbrooke’s Hospital for bMADs], hospital admissions (overnight, emergency), telephone calls (NHS Direct, RSSC helpline, ambulance), use of ‘other’ services (free listing), length of stay in hospital if applicable, diagnostic tests and cause of admission [heart attack, RTA, stroke, ‘other (free listing)’].
At their final visit, patients were asked to rank the three devices and no treatment in order of preference and were allowed to keep their preferred MAD(s). Patients who were intolerant of, or refused, MADs and/or had persistent symptoms at the end of the study were considered for CPAP.
Safety monitoring
Adverse events (AEs) and adverse reactions (ARs) were monitored throughout the trial and recorded at each end of treatment visit. An AE was defined as any untoward occurrence in a clinical investigation subject who was receiving a trial intervention which did not necessarily have a causal relationship with the intervention. An AR was defined as an AE for which a causal relationship with the intervention was at least a reasonable possibility, i.e. the relationship could not be ruled out.
The main expected ARs of MAD therapy were temporomandibular joint/jaw discomfort, mouth discomfort, dry mouth, excessive salivation, gum discomfort, tooth discomfort, loose teeth, malocclusion and mouth ulcers. It was left to the investigator’s clinical judgement whether or not an AE was of sufficient severity to necessitate the patient’s removal from the trial treatment. A patient could voluntarily withdraw from treatment at any time if he or she found an AE to be intolerable.
The severity of AEs was graded as mild, moderate or severe. The relationship between the trial treatment and the AE (the causality) was graded as either unrelated, possibly related, probably related or definitely related by an independent respiratory and sleep medicine consultant physician who sat on the Trial Steering Committee.
All AEs were followed up until resolution or to the end of the AE reporting period.
Serious adverse events (SAEs) were reported to the sponsor within 24 hours of a member of the trial team becoming aware of the event. All SAEs were followed up until resolution or the event was considered stable.
Patient withdrawal
Patients could withdraw from the trial at any time without giving a reason. All patients who withdrew from the study continued to receive normal clinical care if necessary from their GP or consultant in the RSSC.
Sample size and power calculation
Based on the pre-trial systematic review of published studies, the minimum clinically important effect size was considered to be of the order of one-third. An effect size of one-third would be detected with 80% power in a sample size of 72 patients (two-sided significance of 5%). Allowing for 20% loss to follow-up, we aimed to recruit a sample of 90 patients.
Randomisation
Randomisation took place once eligibility was confirmed following measurement for the bMAD and once impression suitability for the SP2 device had been confirmed by the manufacturer. A computer-generated random number sequence produced by the trial statistician determined treatment order. Randomisation was based on two related Williams’ Latin squares designs, with patients randomised in permuted blocks of eight with sequences shown in . Although randomisation in blocks of eight meant that for every eighth patient the sequence was predictable, this was considered to be less important in a crossover trial. Randomisation sequences were held in the research and development (R&D) unit and restricted to research administration staff. The trial team were informed of the randomisation sequence to be given to a patient via telephone contact with the R&D research administrators.
Randomisation sequences according to two Williams’ Latin squares designs
Blinding
Treatment blinding was not possible in this trial. However, the primary outcome, AHI, was ascertained from anonymised PSG traces, which were analysed in batches of 10 by an independent NHS polysomnographer who was not aware of treatment allocations.
Statistical analysis
All patients were followed up irrespective of their level of compliance with the MADs, and all periods for which there was a measurement were included in the analysis using ‘intention to treat’.
Given the nature of the treatments (external devices designed to control symptoms) and the inclusion of a 1-week washout between MAD periods, carryover effects in this crossover trial were considered negligible. In exploratory analysis no treatment by period interactions were identified, which supports this view. Period effects were included in the analysis to account for the long trial period (7–8 months) and in case compliance was related to time in the study.
Initially, the distribution of the outcome measures was assessed by comparing histograms against standard parametric distributions starting with the Gaussian distribution and, if necessary, exploring other plausible families. This was completed for all observations and by treatment group and period. Based on these analyses the primary outcome, AHI, was found to be distributed as a Poisson random variable, which is consistent with a measurement of an event rate per hour. The 4% ODI was also well modelled by a Poisson distribution. All other continuous outcomes were well modelled by Normal distributions. Treatment effects were also plotted over time to further explore period effects.
Given that there were repeated measurements for each patient the main inferential analysis employed a range of mixed models. Initially a full model was fitted that included the main effects of treatment and time period, the interaction between these two and random-effects terms for patient. However, likelihood ratio tests comparing models with and without time by treatment showed that these interaction terms were negligible, and so they were not included in subsequent models. Both treatment and time period in all models were included for consistency and because there was evidence of changes over time in some of the outcome measures based on the likelihood ratio test comparing models with and without the time period effects. The main inferential models were formulated as follows.
For patient i (i =1, . . ., 90) with response yijk for treatment j, j = 1,2,3,4 in time period k, k = 1,2,3,4 we fitted the generalised mixed model,
and
where βo is the intercept fixed at the control treatment in period 1, βj, j = 2,3,4 is the vector of length 3 representing treatment fixed effect, τk, k = 2,3,4 is the vector of length 3 representing the time period fixed effects and μi is the random-effect term for patient i nested in period 1.
For AHI, a Poisson mixed-effects regression was used, with a h( ) log-link function and the random-effects exp(ui) having Gamma(1,α) distribution. A similar Poisson-Gamma model was fitted to the 4% ODI. For both of these models an additional term was included in the regression equation for the times each person was asleep during the test in which the response was recorded. Response to treatment was classified as complete if the AHI was < 5 events/hour, and partial if the AHI was reduced by 50% but was > 5 events/hour; otherwise, patients were classed as non-responders. Mixed-effects logistic regression, using the logit link function, was used to assess treatment effect on complete/partial response, with patient random effects, ui, having a Normal (0,σ2) distribution on the logit scale. All other outcomes were analysed using normal mixed-effects models, with h( ) the identity link function and the ui having a Normal (0,σ2) distribution.
In all analyses estimation of treatment effects was of primary interest, but hypothesis testing was also performed. Nested models were compared using likelihood ratio tests. Model fit was assessed informally by examination of standardised residuals. The approach to multiple testing was as follows. For each of the general(ised) linear mixed models, treatment effects were described as ‘statistically significant’ if the likelihood ratio test comparing the models with and without treatment effects was < 0.05. The TOMADO protocol states that comparison of each MAD against no treatment was important so that, for models that were ‘significant’ overall, we present the significance level is presented based on the Wald test [(βj/se(βj))∼N(0,1)] without adjustment for multiple comparisons. For comparisons between MADs, the (conservative) Bonferroni correction should be applied, that is, standard p-values for these comparisons should be multiplied by 3. Corrections have not been routinely applied, so that readers may make their preferred corrections and where the results are uncorrected has been indicated.
The initial analysis included all patients who completed any treatment period and supplied an outcome measurement. A second analysis included patients who had completed all four periods and provided measurements (complete cases analysis). Both these analyses assumed missing at random for incomplete data and gave almost identical results, so that complete cases results for the AHI and ESS score are omitted from this report. All other results in this report relate to patients who provided any follow-up information. The majority of the missing data arose from patients who did not complete any treatment periods or from sporadic technical failures of the PSG study. These considerations, coupled with (i) the consistency of complete cases and any follow-up analyses, (ii) the consistency of inferences regarding each MAD’s effectiveness across all outcomes and (iii) the clear nature of the results, meant that further sensitivity analysis to account for missing data was considered unnecessary.
Regression analyses were conducted to assess the effects on subsequent AHI and ESS scores of baseline AHI, ESS score, degree of protrusion of the device, age, sex, BMI and compliance, and contemporaneous BMI. These analyses also explored interactions between these variables and treatment effects, although there was limited power. Before the trial, one subgroup analysis of patients who declined CPAP compared with those with mild to moderate OSAH for whom CPAP was not considered necessary. As there were only four patients in the former group, no subgroup analyses were undertaken.
All analyses were performed using Stata (StataCorp LP, College Station, TX, USA) version 12.0 and version 13.0 for Microsoft Windows (64-bit).
Adherence to treatment protocols, treatment preferences, partner scoring assessment, RTAs and AEs were summarised and compared informally. Treatment preference results were available for patients who had completed all four treatment periods and are summarised.
Trial-based economic analysis
The economic evaluation of the crossover trial provided descriptive data on the resource use, unit costs and health state utilities observed during the 4-week periods from the perspective of the NHS.
Resource use
Patient-reported resource use was collected on the case report form (see Appendix 6) for the duration of the trial. Resources used as part of the research protocol that do not affect participant care outcomes (e.g. administering research questionnaires) were omitted. Clinician time required for administering each device was included separately to the reported resource use and priced using the NHS Reference Costs (2011/12)58 for the type of outpatient visit required. Information on medication use during the trial was limited; it was not possible to track start/end date or dosage accurately or to identify which medication usage was associated with which intervention period, therefore they are omitted from the total cost of each intervention. However, medication costs during the trial were negligible.
Unit costs
A NHS supply price was available for SP1 (£18), to which was added the cost of postage (£3) giving a total cost of £21. Instructions were provided with the device for moulding and fitting of the device by the patient and, therefore, no additional clinician time was needed for fitting. As no NHS supply price was available for SP2, the private supply price of £125 was used and, with postage costs of £3, the total cost for SP2 was £128. As the SP1 device can be fitted and managed entirely by the patient, the mould used to manufacture the SP2 is created by the patient using a supplied dental mould kit and, in some cases, patients seek support from a dentist to help with this process. However, in practice, no trial participants required time from dentists to create the SP2 mould.
The bMAD custom device has two significant elements of cost: the manufacture of the custom device itself and two visits to a maxillofacial consultant (for mould creation and fitting). The manufacturer of the custom bMAD provided estimates of the time taken to produce the MAD from the patient’s dental mould (7 hours by a grade 6–8 technician in a NHS maxillofacial laboratory). Using an hourly rate of £50/hour (taken from band 8d of the NHS Agenda for Change pay scales 2011/12) for the technician gave a total cost of manufacture of £350. Materials for production of the bMAD were negligible and, therefore, are considered to be subsumed in the figure of £350. The consultant visits for measurement and fitting of the bMAD were assumed to take a similar amount of time as an average first attendance and follow-up appointment with a consultant at a maxillofacial unit; NHS Reference Costs (2011/12)58 were therefore taken directly. This equated to a cost of £110.36 and £91.95 for the first and second visits, respectively. The total cost of a bMAD was therefore £552.31 (£350.00 + £110.36 + £91.95). If any additional visits to Addenbrooke’s Hospital were required for fitting or measurement, this was recorded on the case report form. The additional visits were priced at the same rate and costs applied in addition to the standard two visits.
As health-care resources and health outcomes were required for a 4-week intervention period, the costs of the MADs were spread over their expected lifetime. For example, as the SP1 and SP2 devices had an expected lifetime of 12 months, the manufacturing costs were multiplied by 4/52 (weeks). Similarly, the bMAD had an expected lifetime of 18 months, so that the costs were multiplied by 4/78 (weeks) for the 4-week intervention period. Point estimates of the life expectancy of devices were provided by the manufacturer but without confidence intervals (CIs). Discussion with the manufacturers indicates that lifetimes may vary around these estimates and this is investigated in the sensitivity analyses. No discount rates are used as a result of the short time horizon of the study.
Unit costs for outpatient care, including labour, capital and overheads, were taken from national estimates.59 The unit costs of any hospital procedures such as outpatient visits or admissions were sourced from the NHS Reference Costs (2011/12).58 In the absence of national estimates, unit costs were taken from published sources60 and centre-specific costs for Papworth Hospital. Appendix 7 shows the unit costs used with sources of data.
In order to inform probabilistic sensitivity analysis, information on the variation of each unit cost (e.g. upper and lower quartiles) was collected and, where no information was available, the standard error (SE) was assumed to be 10% of the mean. For all unit costs, the estimated mean and SEs are assumed to have been generated from a Gamma distribution. All unit costs are valued in 2011/12 British pounds sterling (see Appendix 7).
Unit costs, multiplied by the frequency of resource use, provided a total cost for each item. This was summed by treatment and divided by the number of participants in each intervention group for an average cost per participant by intervention group. The ‘per participant’ resource use costs in Appendix 8 are the raw group means, unadjusted for differences at baseline.
Health state utilities and quality-adjusted life-years
Health state utility weights were taken from two sources: EQ-5D-3L weights were valued using the UK social tariff reflecting the values from a representative sample of the UK population;61 and SF-36 health state responses were converted to the Short Form questionnaire 6-Dimensions (SF-6D) utility scale62 using values from a random sample of the general population in England/UK.63 The utilities are scaled so that full health = 1, death = 0, with the EQ-5D-3L allowing for health states worse than death valued lower than 0 at a minimum of −0.59.
Base-case QALYs use the EQ-5D-3L scores. As the treatment period was a fixed 4-week duration for each intervention and EQ-5D-3L was only collected at one time point for each, the 4-week QALY is calculated as a 4-week proportion of the 52-week year, i.e. QALY = (4 × utility score)/52. The difference in QALYs is not annualised for the within-trial analysis given the short time period.
Methods of cost-effectiveness analysis
The within-trial analysis was a pairwise comparison of mean costs between each treatment and the ‘no-treatment’ control. For each individual and each treatment, total costs were calculated by summing the multiplication of resources used by their unit costs. The ICER was estimated for each MAD against no treatment as the mean of within-patient difference in total 4-week costs, divided by the within-patient difference in 4-week QALYs. A mixed-effects model was used to estimate within-patient differences in total costs and QALYs. Differences in costs and differences in QALYs were estimated in separate models. Baseline EQ-5D-3L scores, patient weight and the time period, were included as covariates. In addition, for comparisons between each treatment, the incremental net monetary benefit (INMB) over 4 weeks was estimated assuming that decision-makers are willing to pay £20,000 per QALY.
Probabilistic sensitivity analysis was conducted to incorporate the uncertainty in estimates cost and effects. Samples (with replacement) of patients were generated and for each sample the mixed-effect model was rerun and unit costs were resampled from the estimated Gamma distributions. Two thousand bootstrap samples produced a set of possible costs and effects for each intervention, each of which were used to estimate an incremental cost (difference in total cost) and incremental effect (difference in QALYs). These were used to construct a series of cost-effectiveness acceptability curves (CEACs) which plot the probability that each MAD is cost-effective against the maximum WTP for one QALY. In addition, a cost-effectiveness acceptability frontier (CEAF) was constructed to plot the most cost-effective device against the maximum WTP.
Deterministic sensitivity analyses were conducted to assess the impact on the INMB of changes in the purchase price of each MAD and varying the expected lifespan of devices from 6 to 60 months. Assumptions regarding rare events and complications, such as RTA, were investigated in the sensitivity analyses in the long-term model of cost-effectiveness (see Chapter 4).
The Trial of Oral Mandibular Advancement Devices for Obstructive sleep apnoea–hypopnoea results
Patient recruitment
Between December 2010 and July 2012, 440 patients were screened for the trial. Two hundred and eighty-one patients were excluded at screening, 51 of whom were excluded for dental ineligibility by a sleep physician. A total of 159 patients gave written informed consent. Sixty-nine patients either refused or were ineligible following the baseline sleep study or the hospital visit for the bMAD fitting. Only two patients who were considered dentally suitable for the trial by a sleep physician were subsequently excluded by the hospital maxillofacial team for poor oral hygiene and tooth decay. The remaining 90 patients were recruited to the trial and received a randomised treatment allocation sequence ().
Patient flow through the trial.
Baseline characteristics
Baseline measurements are recorded in . Mean (SD) age was 50.9 (11.6) years and ranged from 26 years to 79 years. Eighty per cent were men (72/90). Mean (SD) AHI at baseline was 13.8 (6.2) events per hour, with three patients who were accepted on the basis of desaturation index (DI) having a baseline AHI of < 5 events per hour, rendering them ineligible for the trial on confirmatory PSG. These patients were retained in the trial according to ‘intention to treat’. Mean (SD) ESS score was 11.9 (3.5) and, although 12 patients had a baseline ESS score below the acceptance threshold of 9, they were eligible based on an ESS score of ≥ 9 at screening. Again these patients remained in the trial.
Baseline characteristics of trial patients
Risk factors for heart disease were common in this group. Median [interquartile range (IQR)] BMI was 30.6 kg/m2 (27.9–35.1 kg/m2) and mean BP pressure was normal at 130/80 mmHg, but varied widely from 98/57 to 177/116 mmHg. Diabetes was present in eight (9%) patients, 23 (26%) were being treated for hypertension and 21 (23%) for hypercholesterolaemia. Five (6%) patients had been diagnosed with ischaemic heart disease and three (3%) had previous cardiovascular events (CVEs).
Of the 90 patients entered into the trial, 86 were new patients (who had not refused CPAP) and four were patients who had tried CPAP but could not tolerate it.
Withdrawals
shows patient progress through the trial. During the trial, 16 (18%) patients withdrew and the reasons for withdrawal are described in the .
Characteristics of patients who withdrew during the study
Of the 16 patients who withdrew from the trial, seven (8%) did not complete any treatment periods, three were using the bMAD, two were using the SP1, one was using the SP2 and one patient was in the no-treatment arm. A further two (2%) patients who withdrew between the first and second treatments provided no primary outcome data as a result of technical failure of the sleep study after the first treatment period. These nine patients (7 + 2) provided no information after baseline and are excluded from all analyses. The main reasons for withdrawal in patients who did not complete any treatment period were intolerance of a device or were related to an AE. It is likely that these patients would not tolerate any of the devices and all wanted to try alternative treatments (CPAP or CM including weight loss).
Seven (8%) further patients withdrew during the trial: four were using the bMAD, one was using SP1 and two were using the SP2. Only one of these withdrawals (SP2) was as a result of intolerance to the device. These cases were included in the main analysis. Seven other sleep studies failed, leaving 305 studies (85% of 360) in 81 patients [of 90 (90%)] for AHI analysis. For all other outcomes, 314 (87%) measurements and 83 (92%) patients were available for analyses.
One patient who was randomised early in the trial was unable to remould another SP2 to replace their damaged SP2 and subsequently withdrew. Thereafter successful SP2 moulding was made a prerequisite for randomisation. Four patients were subsequently not randomised because they could not mould the SP2. This was in part because of intolerance that would probably have applied to all three devices, but technical difficulty was a factor in some cases.
Baseline characteristics for the patients who withdrew from the study were similar to those who completed follow-up (data available on request).
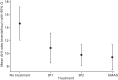
Primary outcome: apnoea–hypopnoea index
shows the mean AHI (SD) for each treatment, alongside the results of the Poisson-gamma regression analysis. Mean AHI for each treatment is plotted in . This shows that that the rate of apnoea/hypopnoea events per hour for each MAD, relative to no treatment, is reduced significantly, with estimated relative rates of 0.74, 0.69 and 0.64 for SP1, SP2 and bMAD, respectively. The reductions for the SP1, SP2 and bMAD represent effect sizes of approximately 0.36, 0.47 and 0.49 SDs, respectively, all of which exceed the minimum clinically important difference of one-third proposed during study planning. In post-hoc pairwise comparisons there were no significant differences in AHI between the different MADs ().
Summary of results from mixed-effects model for AHI (n = 81)
Estimated mean AHI and 95% CI for the four treatments from the Poisson-Gamma model.
Comparison of AHI between different MAD
Examination of the standardised residuals for AHI showed that this model was a good fit to the data, with no systematic effects observed.
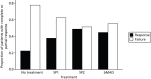
Apnoea–hypopnoea index: responders to treatment
Of the patients who had an AHI value for at least one treatment, complete or partial AHI response during MAD use was observed in 29 (38%) patients for the SP1, 38 (49%) patients for the SP2 and 33 (45%) patients for bMAD, compared with 17 (22%) patients during the no-treatment period ( and ). Patients who responded to one MAD were more likely to respond to others, but this was not completely predictable. Four of the 74 completers (5%) had a complete response to all treatments, nine (12%) had a partial or complete response to all treatments and 20 (27%) did not have a response to any treatment. The four patients who completely responded to all treatments also had low AHI during the no-treatment period (AHI at baseline 3.1, 5.4, 7.6 and 8.9).
Response of patients by treatment
Complete or partial response of patients by treatment.
Predictors of apnoea–hypopnoea index response
Using mixed-effects logistic models for complete/partial response, all MADs had significantly greater response rates than during the no-treatment period (). Response was significantly associated with baseline BMI [odds ratio (OR) 0.89, 95% CI 0.81 to 0.98; p = 0.014] and with contemporaneous BMI (OR per kg/m2 0.88, 95% CI 0.80 to 0.98; p = 0.007). It was also weakly associated with protrusion (OR 1.03 per % protrusion, 95% CI 1.00 to 1.05 per % protrusion; p = 0.034). Baseline AHI, ESS score, sex and age (years), as well as measures of compliance, were not significantly associated with response.
Summary of results from mixed-effects logistic regression for complete or partial response to treatment (n = 81)
Secondary outcomes
Epworth Sleepiness Scale
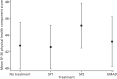
shows summary statistics for the four treatment periods as well as the results of the mixed-effects linear regression. plots estimated ESS score by treatment. There was a clear, statistically significant, reduction (improvement) in ESS score for all MADs compared with no treatment, with effect sizes of approximately 0.35, 0.50 and 0.55 SDs compared with no treatment. In addition, there was a weakly significant difference between the SP1 and the bMAD in post-hoc pairwise comparisons ().
Summary of results from mixed-effects model for ESS score (n = 83)
Estimated mean ESS score and 95% CI for the different treatments from the mixed-effects model.
Comparison of ESS score between different MADs
Four per cent oxygen desaturation index
The findings for 4% ODI mirrored those for AHI, as can be seen in . Although all MADs used resulted in significantly lower desaturation index relative to no treatment, there were no significant differences between MADs. In general, similar patterns were observed for minimum and mean SpO2 and time with < 90% SpO2 (data available on request).
Summary of results from mixed-effects model for 4% ODI (n = 81)
Daytime blood pressure
Blood pressure was taken three times at each visit and the average of the three measurements recorded. There was very little evidence of an effect of any of the MADs on either SBP or DBP during the trial. Mean (SD) SBP and DBP at the end of the no-treatment period was 127.4 mmHg (12.2) and 79.2 mmHg (8.3), respectively. For SBP, the mean (SD) at the end of treatment with the SP1, SP2 and bMAD was 127.0 mmHg (13.5), 128.8 mmHg (14.7) and 127.2 mmHg (12.6), respectively. Corresponding results for DBP were 79.0 mmHg (9.4), 79.9 mmHg (9.2) and 79.5 mmHg (10.0), respectively.
Treatment compliance
Of the 314 sleep diaries expected from the 81 patients who completed at least one period, 14 were not returned and three were not completed satisfactorily. Compliance was slightly worse in terms of the number of nights used, and significantly worse for duration of use per night, for the SP1 than for the SP2 or the bMAD (; p < 0.001), but there were no significant differences in compliance between the SP2 and the bMAD. Patients were also more likely to discontinue use of the SP1 than the other two devices ().
Compliance with treatment
Treatment interruption or discontinuation for patients who used the device for < 28 days
Patient evaluation of treatments
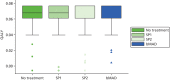
On average, patients considered the SP2 and the bMAD to be as comfortable as no treatment, but the SP1 was significantly less comfortable than all other treatments (VAS for comfort, ). This resulted in greater satisfaction for the SP2 and the bMAD than for no treatment or the SP1 (VAS for satisfaction, ). shows that patients reported that the SP1 was more likely to fall out during sleep than the SP2, and that the SP2 was more likely to fall out than the bMAD. In addition, patients reported that they were more likely to remove the SP1 during sleep than either the SP2 or the bMAD ().
Summaries of the visual analogue valuations of treatment comfort and satisfaction
Patient report of frequency that device fell out
Patient report of frequency that device was removed
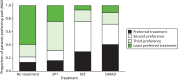
The 74 patients who completed all treatments were asked to state their preferred treatment. Of these, 30 (41%) ranked the bMAD highest and 23 (31%) ranked it second. The SP2 was ranked highest by 22 (30%) patients and second by 34 (46%) (). Only 10 (14%) patients ranked no treatment highest.
Bar chart of patient preference.
Most patients (56/90, 62%) continued with their preferred device after the study ended, with five (6%) others retaining the MAD that gave the best results for them. Other treatments undertaken by patients after the trial are listed in .
Patient management after completing TOMADO
Functional Outcomes of Sleep Questionnaire
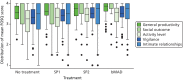
Eighty-three (92%) patients had at least one FOSQ result (). summarises the results for the five subscales. The overall FOSQ scores showed a weak period effect (p = 0.021), suggesting that there may be some adjustment of questionnaire responses over time. After including period effects in the model, there were significant improvements for all the MADs compared with the no-treatment period. In addition, there were small but significant differences between the SP1 and SP2 and between the SP1 and bMAD but not between the SP2 and bMAD (). The plot of individual FOSQ scales () suggests that this improvement is because of small increases in all dimensions but particularly for activity level and general productivity.
Summary of results from mixed-effects model for the FOSQ (n = 83)
Estimated mean FOSQ and 95% CI for the different treatments from the linear mixed-effects model.
Comparison of total FOSQ score between different MADs
Box plots of the mean score for each domain of the FOSQ.
Short Calgary Sleep Apnoea Quality of Life Index
The summaries and model results for the overall score are shown in and . In common with the FOSQ overall score, there was a significant effect of all MADs compared with no treatment and a small but significant difference between the SP1 and SP2 and between the SP1 and bMAD, but not between the SP2 and bMAD (). shows results for each subscale of the SAQLI and, again, shows a small improvement across all dimensions, particularly daily activities and symptoms.
Summary of results from mixed-effects model for the SAQLI (n = 83)
Estimated mean SAQLI score and 95% CI for the different treatments from the linear mixed-effects model.
Comparison of total SAQLI score between different MADs
Box plots of the mean score for each domain of the SAQLI.
Short Form questionnaire-36 items
Summaries of results for the SF-36 standardised PCS and MCS are shown in and and . Predictably, this general HRQoL instrument is less sensitive to differences between the treatments than the disease-specific instruments, with only the comparison between the SP1 and SP2 showing a borderline significant difference in PCS in favour of the SP2. There was a similar borderline significant increase in the MCS for the bMAD compared with the SP1.
Summary of results from mixed-effects model for the SF-36 standardised PCS and MCS (n = 83)
Standardised SF-36 physical health summary.
Standardised SF-36 mental health summary.
Protrusion achieved
The protrusion achieved was measured for all three devices at Papworth Hospital (). The SP1 achieved the greatest protrusion, being 0.89 mm (95% CI 0.42 to 1.37 mm; p < 0.001) greater than the SP2 and 0.66 mm (95% CI 0.17 to 1.14 mm; p = 0.008) greater than the bMAD. In a model that contained the degree of protrusion, MAD and time period, protrusion did not influence AHI [hazard ratio (HR) 0.997, 95% CI 0.991 to 1.001; p = 0. 206]. Protrusion did have a small effect on the probability of a response to treatment (see earlier section on predictors of response to treatment).
Learning effect
The SP1 and SP2 devices were moulded and protruded by patients independently; in contrast, in the case of the bMADs, protrusion was determined by a medical professional. Variability between the mean protrusion values for each device may have been the result of a variety of factors, including patient-determined compared with clinician-determined protrusion and previous experience of wearing a device on the trial. For example, the SP1 was moulded at the start of that treatment period. Therefore, in patients with SP1 as their second or third device, jaw protrusion may have been either more or less depending on acclimatisation to jaw protrusion and any positive or negative effects experienced while using previous devices. A few patients may have been unintentionally guided by their experience of the bMAD-fitting process. Six patients commented that they had found the visit to the maxillofacial team for bMAD fitting useful in subsequently informing SP2 moulding, including protrusion. Two patients commented that the bMAD-fitting experience helped when they later moulded the SP1. Two others did not describe inadvertent dental guidance, but found the SP1 easier to fit having already made the SP2 mould.
Safety reporting
Driving
Eighty-seven (97%) patients in TOMADO reported that they were drivers at baseline and three (3%) were not. Eighty-six patients drove a car, two a motorbike, three a heavy goods vehicle and 16 drove other vehicles including a fork lift truck, jeep, van, minibus and tractor. records patient-reported sleepiness while driving. There was a clear improvement in sleepiness while driving, and in the requirement for interruption to journeys, during all periods of MAD use compared with no treatment, but little difference between MADs. During the trial there were only three reported cases of ‘nodding off’ (none of which resulted in a collision) and five collisions while driving. No collisions resulted in an injury to anyone involved other than the patient, and one collision resulted in an injury to the patient, who required treatment and advice from a health-care professional.
Patient-reported sleepiness associated with driving
Partner-evaluated snoring scale
Fifty sleeping partners of trial patients completed the snoring VAS for all four periods ( and and ). This showed a clear improvement for all MADs compared with no treatment, and between the SP1 and the two more sophisticated devices.
Summary of effects from mixed-effects model for the partner-rated VAS for snoring (n = 50)
Comparison of the partner-rated snoring scale between different MADs
Partner-evaluated snoring scale.
Adverse events
There were four SAEs during the trial. There was one case of sick sinus syndrome with atrial flutter and one case of hypoglycaemia during periods of no treatment, both considered possibly related to OSAH, one case of complete heart block and one case of non-specific chest pain during bMAD use, both considered possibly related to OSAH and MAD use. These occurred in four separate patients and all events were resolved within 7 days.
A total of 851 minor AEs were recorded in 86 patients who enrolled in the trial (). These were mainly mouth discomfort and excess salivation. They were recorded equally frequently for all three MADs and less frequently during the no-treatment periods. Among patients who withdrew from the study, there were 63 AEs in 12 patients, mainly mouth discomfort (52, 83%). Almost all minor AEs in both completers and withdrawals were classed as probably related to MADs (528 events in 85 patients) or possibly related to both OSAH and MAD use (174 events in 54 patients) by an independent sleep physician.
All reported AEs during the trial with number of patients affected in brackets
Specific events included in each category are given in Appendix 9.
Trial-based economic analysis
Data completeness
Data were formatted as a four-period (n = 83) observation panel, including participants with at least one completed treatment period and for whom complete data on QoL and resource use were available. Of the 83 people, 77 provided complete EQ-5D-3L and resource use data for the SP1, SP2 and control periods, and 75 for the bMAD and control periods.
Seventy-four participants provided complete EQ-5D-3L and resource use data for all intervention periods. Data completeness was similar for the SF-6D (n = 76 for SP1, SP2 and control, and n = 76 for bMAD and control period).
Costs
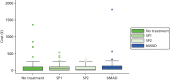
shows that the SP1 device cost the least (£1.62) pro rata over the 4-week trial period, followed by SP2 (£9.85), and that the bMAD is considerably more expensive (£28.64). The mean non-device costs during the no-treatment period were £78.50, while they were £73.02 for SP1, £53.58 for SP2 and £76.25 for bMAD (). shows box plots of total costs for each group. While costs were similarly clustered for each trial group, SP2 had the narrowest spread of cost. The bunching of outliers close to the upper quartile tend to comprise patients with more frequent primary care (e.g. to dentist or GP) or outpatient visits and these occurred in all groups. However, in both the control and bMAD groups, a few patients incurred very high costs as a result of rare events such as an atrial flutter, pacemaker implantation and hypertension with chest pain.
Trial-based comparison on costs incurred over 4 weeks
Box plots of total cost during each 4-week treatment period.
Combining the device and resource use costs and comparing each intervention with control over the 4-week intervention period shows that the SP1 compared with control was £4 less (SE £21), and that SP2 was £15 less on average (SE £21), but that the mean cost of bMAD was £26 greater than mean costs in the control group (SE £28) (). Differences were not statistically significant.
Quality-adjusted life-years
and show the distribution of the QALY scores at baseline and by treatment group using the EQ-5D-3L and SF-6D. The EQ-5D-3L shows that the SP2 and bMAD have better profiles, with more people scoring around 0.078 or above and bMAD also having fewer people with scores around zero. Of those people with low outlying QALY scores, one person had consistently low scores during baseline and all four intervention periods and another at baseline and three treatment periods. The remaining differences show two participants with QALYs outside the lower IQR while on the SP2 and bMAD, and one participant during the no-treatment period. In only one case did the participants who reported lower EQ-5D-3L QALY scores also accrue higher costs, and this was during the no-treatment phase.
Box plot of EQ-5D-3L QALY results by treatment.
Box plot of SF-6D QALY results by treatment.
The mean QALY score based on the EQ-5D-3L data for the control period was 0.065 (SE 0.002). To give some perspective, 4 weeks in perfect health is associated with a QALY score of (1 × 4)/52 = 0.0769. The control score is, therefore, less than perfect health, equating to a QALY score of 0.065. The difference in EQ-5D-3L QALY values for each MAD compared with no treatment (see Appendices 10 and 11) was 0.0009 (SE 0.001) for SP1, 0.0009 (SE 0.001) for SP2 and 0.0018 (SE 0.001) for bMAD (see ). Although the gain was greatest for bMAD, there was substantial uncertainty, shown by the large SEs. The 95% CI for the effectiveness of each device compared with control spanned zero (i.e. no statistically significant effect).
Trial-based comparison of costs and QALYs from devices against control
The SF-6D showed that the SP2 conferred the best health outcomes, with mean QALY score around 0.057, followed by bMAD and no treatment, around 0.053, and SP1, around 0.052. The one participant recorded as an outlier using SF-6D QALYs had fewer QALYs on no treatment, SP2 and bMAD. As with the EQ-5D-3L QALYs, those with low outlying QALY values were not necessarily those with higher costs. The mean QALY score from the mixed-effects model for the control period was 0.053 (SE 0.0008). The difference in SF-6D QALYs (see Appendices 11 and 12) compared with no treatment during the 4-week intervention period was 0.0004 (SE 0.0008) for the SP1, 0.0019 (SE 0.0007) for SP2 and 0.0009 (SE 0.0009) for the bMAD. Of all the MADs, the SP2 showed the greatest change in QALYs compared with control and was also the only intervention with a statistically significant difference (p = 0.013).
Cost-effectiveness
shows that the ICERs were negative for the SP1 and SP2 compared with control, i.e. costs were lower and outcomes better for the two interventions than for no treatment. Note that EQ-5D-3L QALY differences between devices were small and non-significant. Of these two, the SP2 is more beneficial as costs were lower than the SP1.
also shows that bMADs have the greatest impact on QALY gain, and at a cost of £14,900 per additional QALY gained, would be considered a cost-effective treatment compared with control. However, compared with the SP2, the bMAD costs an additional £46,000 per QALY (£105 – £64)/(0.0667 – 0.0658 QALYs). These results are mirrored by the net monetary benefit, which shows that the SP2 achieved the highest INMB, compared with no treatment, at £33 per 4 weeks assuming a WTP of £20,000 per QALY ().
The uncertainty around estimates of cost per QALY gained is represented in the cost-effectiveness planes (–) and CEAF (). These indicate the results are robust. The CEAF () shows the SP2 to be most cost-effective up to a WTP per QALY of £39,800, at which point the bMAD supersedes it (39% likelihood of being cost-effective compared with 35% for the SP2). Below a WTP of £5000 per QALY only SP2 is more cost-effective than no treatment. Deterministic sensitivity analyses also showed that results are robust to using only complete case analysis as well as changes in a device’s price and lifespan (see Appendix 13, –39). When the bMAD price exceeds £525 or average lifespan falls < 14 months, it no longer has a positive INMB. When the price of the bMAD falls to below £60, or its length of life extends to beyond 3 years (with no change in the SP1), it becomes more cost-effective than the SP1. However, even when assuming the same price for the bMAD of £60 or that its lifetime is at least 5 years, the bMAD remains less cost-effective than the SP2.
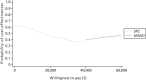
Incremental cost-effectiveness plane: SP1 compared with no treatment.
Incremental cost-effectiveness plane: bMAD compared with no treatment.
Cost-effectiveness acceptability frontier for each MAD compared with no treatment.
Incremental cost-effectiveness plane: SP2 compared with no treatment.
The cost-effectiveness analysis was repeated using the SF-6D data for health outcomes. Compared with no treatment, the SP1 has a QALY gain of 0.0004 (SE 0.0007) with the same cost saving described above (−£4 vs. control), meaning the SP1 was both cheaper and more effective, dominating no treatment. However, neither the difference in costs compared with no treatment nor the difference in health outcomes was statistically significant. The SP2 had a statistically significant improvement in health outcomes when compared with no treatment of 0.0019 (SE 0.0007) QALYs, with a p-value equal to 0.013. Combined with the costs saving of £15 over 4 weeks compared with no treatment, showing the SP2 to be dominant over no treatment; being both cheaper and more effective than no treatment. The bMAD provided an improvement in health outcomes compared with no treatment of 0.0009 (SE 0.0009) QALYs, although this was not statistically significant. The bMAD cost £26 more than the no-treatment control, giving an ICER of £30,743 per QALY.
Applying a WTP per QALY of £20,000 the INMB of each treatment compared with control was calculated. The INMB for the SP1 was £12, for the SP2 £52 and for the bMAD £9. Probabilistic sensitivity analysis was used to produce the CEAC and CEAF based on the SF-6D results (Appendix 13, ) representing the uncertainty in costs and QALY estimates. This analysis found the SP2 to have the highest probability of being the most cost-effective treatment at all WTP thresholds per QALY. Above a WTP of £20,000, the SP2 had a probability of being the most cost-effective in excess of 95% compared with the SP1, bMAD or no-treatment alternatives.