

Pathogen Detection Help Document
Beta Release
This is a beta release of the Pathogens help documentation in order to make new content available, while development continues on the format and presentation of the information. Navigation tips:
pd-help@ncbi.nlm.nih.gov. |
Table of contents
- What is the NCBI Pathogen Detection project?
- How To
- Pathogens Project Components (resources/tools, types of data, contributors)
- Data Retrieval & Analysis
- Isolates Browser help
- SNP Tree Viewer help
- Automatic e-mail notifications of new data
- Antimicrobial Resistance (AMR) resources
- Overview (schematic illustration of AMR resources)
- MicroBIGG-E: Microbial Browser for Identification of Genetic and Genomic Elements (search tips, data fields, use cases/sample searches)
- MicroBIGG-E Map
- Pathogen Detection Reference Gene Catalog (search tips, data fields, use cases/sample searches)
- Pathogen Detection Reference HMM Catalog (search tips, data fields)
- Pathogen Detection Reference Gene Hierarchy (search tips, data fields)
- AMRFinderPlus
- FTP Site help
- Data Submissions
- Data Processing Pipeline
- Data Retention and History Tracking
- Log of Changes to the Pathogen Detection Project
- References
- Contact the NCBI Pathogens Detection Team
What is the NCBI Pathogen Detection project? 

- Overview
- Where to access the Pathogens Detection Project results
- Where to access the Pathogen Detection Project Antimicrobial Resistance (AMR) Data
- Update Frequency
- References and Contact Information
Overview 
NCBI Pathogen Detection integrates bacterial and fungal pathogen genomic sequences from numerous ongoing surveillance and research efforts whose sources include food, environmental sources such as water or production facilities, and patient samples. Foodborne, hospital-acquired, and other clinically infectious pathogens are included. The system provides two major automated real-time analyses:
- It quickly clusters related pathogen genome sequences to identify potential transmission chains, helping public health scientists investigate disease outbreaks
- As part of the National Database of Antibiotic Resistant Organisms (NDARO), NCBI screens genomic sequences using AMRFinderPlus to identify the antimicrobial resistance, stress response, and virulence genes found in bacterial genomic sequences, which enables scientists to track the spread of resistance genes and to understand the relationships among antimicrobial resistance, stress response, and virulence.
NOTE: NCBI Pathogen Detection does not identify outbreaks or outbreak membership. All analyses are dependent on the public data submitted to the system and the quirks of our analysis pipelines. NCBI provides a service to help identify clonal relationships based on genomic similarity. Determinations of outbreaks are done by public health organizations including CDC, FDA, USDA. Although we take care to make the analyses as error free as possible, this is a large-scale automated pipeline that takes data from submitters and analyzes it in real-time therefore we cannot guarantee the results to be free from error or applicable for a particular use.
Where to access the Pathogens Detection Project results
- Pathogens Detection home page - provides an overview of the project and links to pathogens resources and tools.
- Pathogen Detection Isolates Browser - provides an interface to search and/or subset the isolate data, displays details for each isolate, and links to a SNP Tree Viewer, which shows phylogenetic relationships among the isolates. (Separate sections of this file provide Isolates Browser help documentation and SNP Tree Viewer help documentation.)
- Microbial Browser for Genetic and Genomic Elements (MicroBIGG-E) - Provides a detailed view of genetic elements important to clinical and public health identified by AMRFinderPlus. See the MicroBIGG-E documentation for more information.
- The Pathogen Detection Reference Gene Catalog, Reference Gene Hierarchy, and Reference HMM Catalog are the reference databases behind MicroBIGG-E and together they make up the AMRFinderPlus database. See Antimicrobial Resistance (AMR) Resources for more information.
- FTP site provides access to the results of analyses that have been done at NCBI on the sequence reads and genome sequences of pathogen isolates. (A separate section of this document provides an overview of the data available on the FTP site, and the FTP readme file provides additional details.)
Where to access Antimicrobial Resistance (AMR) Data
- MicroBIGG-E is the Microbial Browser for Identification of Genetic and Genomic Elements. Every row in the MicroBIGG-E display is an anti-microbial resistance (AMR), stress response, and/or virulence gene that has been identified in an isolate by the data processing pipeline.(Separate sections of this file provide MicroBIGG-E help documentation.)
- AMR Landing page - provides information about the NCBI National Database of Antibiotic Resistant Organisms (NDARO), a collaborative, cross-agency, centralized hub for researchers to access AMR data to facilitate real-time surveillance of pathogenic organisms.
- AMR Resources page - provides a list of available resources, with a brief description and sample searches or links to additional information about each one.
- Pathogen Detection Reference Gene Catalog provides access to a curated reference set of antimicrobial resistance genes and proteins, which are stored in the Bacterial Antimicrobial Resistance Reference Gene Database (BioProject PRJNA313047). The Reference Gene Catalog together with the Reference Gene Hierarchy and the Reference HMM Catalog make up the AMRFinderPlus database and provide the reference data behind the AMRFinderPlus software and MicroBIGG-E browser. The source of input for these curated databases include: 1) allele assignments, 2) exchanges with other external curated resources, 3) reports of novel antimicrobial resistance proteins in the literature. (The Pathogen Detection Reference Gene Catalog, Pathogen Detection Reference Gene Hierarchy, and Pathogen Detection Reference HMM Catalog help documents provide information on how to use the tools.)
- AMRFinderPlus - a tool that compares isolate genomes against the reference protein set using BLAST and against the HMM set using HMMER, and uses the gene hierarchy to provide the most specific protein assignment to antimicrobial resistant protein or family, if present in the query set of proteins. AMRFinderPlus identifies the AMR genes and point mutations that are found by the original AMRFinder, plus it identifies select members of additional classes of genes such as virulence factors, biocide, heat, acid, and metal resistance genes. Unlike other AMR gene detection methods that report the best hit, AMRFinderPlus reports the specific gene symbol based on the available evidence. For example, when presented with a novel blaKPC allele that is nearly identical to blaKPC-2, closest hit tools might return blaKPC-2, but AMRFinderPlus would call it as blaKPC so that users do not incorrectly assume the phenotype (illustrated example). More details about the tool are provided in publications Feldgarden M, et al., 2019 and Feldgarden M, et al., 2021.
- Submit sequence and phenotype data related to AMR - includes instructions on how to submit data for real-time analysis, submit antibiograms to the BioSample database, and request new alleles for beta-lactamase, MCR, and Qnr Genes.
- FTP/Raw Data Download - includes AMRFinderPlus data files, the Reference Gene Catalog, Reference Gene Hieararchy, and Reference HMM Hierarchy, and the Bacterial Antimicrobial Resistance Reference Gene Database (BioProject PRJNA313047) See database documentation for more information about the files and formats.
Update Frequency
- FTP Results and Isolates Browser and MicroBIGG-E
- The Pathogens Detection Project analysis results are updated approximately daily for each taxgroup, when new data is submitted. FTP Results and the Isolates Browser are generally synchronized, but sometimes indexing delays might occur, resulting in a temporary asynchrony. The Isolates Browser and MicroBIGG-E updates are synchronized and update along with the corresponding taxgroups. The Isolates and MicroBIGG-E tables in Google Cloud BigQuery are updated daily and data may lag behind the web interfaces by up to one day.
(Read an overview of the FTP site, or go directly to the FTP parent directory or to the FTP Results subdirectory. Read about the Isolates Browser.) - Isolate Browser and MicroBIGG-E data in Google Cloud are updated independently, see GCP Update frequency for details.
- The Pathogens Detection Project analysis results are updated approximately daily for each taxgroup, when new data is submitted. FTP Results and the Isolates Browser are generally synchronized, but sometimes indexing delays might occur, resulting in a temporary asynchrony. The Isolates Browser and MicroBIGG-E updates are synchronized and update along with the corresponding taxgroups. The Isolates and MicroBIGG-E tables in Google Cloud BigQuery are updated daily and data may lag behind the web interfaces by up to one day.
- The AMRFinderPlus database (Including the Reference Gene Catalog, Reference Gene Hierarchy, Reference HMM Catalog, and AMRFinderPlus database are updated simultaneously, somewhat irregularly with releases approximately every two months.)
- FTP Rapid Reports
- The Rapid Reports directory of the Pathogens FTP site is a pilot phase test of rapid reporting based solely on wgMLST allele differences and currently reports on certain bioprojects and some organisms. The FTP Rapid Reports for a given organism are updated within approximatly an hour of completing sequence read submissions for a new isolate.
(Read an overview of the FTP site, or go directly to the FTP Results subdirectory, where you can find Rapid Reports for organisms such as Clostridioides difficile, Salmonella, Listeria.)
- The Rapid Reports directory of the Pathogens FTP site is a pilot phase test of rapid reporting based solely on wgMLST allele differences and currently reports on certain bioprojects and some organisms. The FTP Rapid Reports for a given organism are updated within approximatly an hour of completing sequence read submissions for a new isolate.
References and Contact Information
- References about the Pathogen Detection Project and related intiatives
- Contact information for the NCBI Pathogens Detection Team
How To:
- Visual HowTos
- Quick link searches (e.g., quickly retrieve new isolates for a given organism group)
- General text searches (e.g., "lettuce")
- Field-specific searches (e.g., retrieve isolates that were collected in a given geographic location, e.g., U.S.)
- Using filters to focus the search results (e.g., retrieve isolates by scientific name, then facet the data by various criteria such as isolation source)
- Identify the possible source of an outbreak (e.g., E. coli outbreak from all-purpose flour)
- The Advanced Search > Examples of SOLR queries provides additional examples of field-specific searches and complex Boolean searches using the SOLR query syntax
- The Pathogen Detection Reference Gene Catalog > Use cases/sample searches section of this document shows how to find antimicrobial resistance (AMR) genes, point mutations that confer resistance, AMR content of known isolates, and more.
Quick link searches
- The Pathogen Detection Project home page includes an "Explore the Data" section. This lists the four foodborne pathogens including direct links to the Isolates Browser for Salmonella enterica, E.coli and Shigella, Campylobacter jejuni, Listeria monocytogenes, and provides instant access to isolates from those groups.
- The Organism Groups page also provides links for all available organism groups, along with additional details for each group. Note that the species name under the Organism Groups table reflects the most common species in each group, but does not reflect all species. For example, the Salmonella enterica organism group consists of predominantly Salmonella enterica isolates, but also Salmonella bongori isolates. To see the full list of organisms present in each group, see the scientific_name column in the Isolates Browser.
- For example, to quickly retrieve new isolates for a Salmonella enterica, open the Pathogen Detection Project home page:
- Scroll down to "Explore the Data" and follow the "New Isolates" link for the Salmonella enterica.
- That will retrieve isolates that have become available in the Pathogen Detection Project. "New" isolates are those that have been added to a Pathogen Detection Group (PDG#) since the last calculation. This may have been all isolates added in the last 24 hours for frequently updated organism groups like Salmonella, or it may have been months since the last update and "new" isolates are now several months old, but reflect the newest isolates added to a given pathogen detection group.
General text searches
- Simply enter text term(s) of interest (e.g., "lettuce") in the Isolates Browser to search across all of the text-containing fields.
- For example, open the Isolates Browser home page.
- It will display all isolates are by default. Any search you enter will result in the display of only the subset of isolates that match your query.
- Enter the desired search term in the text box to display of only the subset of isolates that match your query. For example, try searches for:
- lettuce
- strawberries
- "all-purpose flour"
(Separate sections of this document describe how the Isolates Browser handles special characters such as hyphens that are part of search terms, and provide tips about case sensitive searches and the use of quotes for phrase searches.) - blaKPC*
(Separate sections of this document provide additional examples of searches for antimicrobial resistance (AMR) genotypes and details about the use of wildcards such as asterisks.) - Use the Filters, if desired, to filter the data you retrieved by characteristics such as:
- geographic location where the isolate was collected
- isolation type (clinical or environmental/other)
- property (has AMR genotypes or has AST phenotypes)
- target creation (date on which the isolate was first seen at the Pathogen Detection project)
- and more...
- For additional information, search tips, and examples, see the Isolates Browser help > Allowable search terms section of this document.
Field-specific searches
- As an alternative to general text searches, you can conduct more precise searches by limiting your query to specific data fields.
The general syntax of a field-specific search is:
- searchfieldname:searchterm (to search for a single term)
- searchfieldname:"search phrase" (use quotes to search for a phrase)
- searchfieldname:searchterm AND searchfieldname:searchterm (use the desired Boolean operator(s))
Important notes:
- The names of data fields, and the values they contain, are case sensitive.
- The exact name of the data fields can be seen by hovering the mouse over the column names, then a popup appears with the search syntax for that field.
- The data field names and values might also include special characters such as underscore bars, hypens, parentheses, and slashes. These should be included in the query string, as the Isolates Browser has been modified relative to the SOLR Standard Query Parser to recognize and properly handle special characters that are part of a search term.
- For example, you can search the Location data field, as shown below, in order to retrieve isolates that were collected from a given geographic area:
- Open the Isolates Browser home page. It will display all isolates are by default.
- Enter the following type of search in the text box to display only the subset of isolates that have been identified by the submitter as having been collected in the USA:
- geo_loc_name:USA
- For additional examples, such as searches that retrieve isolates with specific genotypes and/or phenotypes, see the Examples of SOLR queries section of this document.
- For detailed information about searching specific data fields, see the Isolates Browser help > Advanced Search > Data Fields section of this document.
Using Filters to focus the search results
- You can use "Filters" in order to focus on a specific subset of isolates.
- For example, open the Isolates Browser home page. It will display all isolates by default.
- To filter the isolates by criteria such as isolation source:
- Click on the "Filters" menu in order to filter the data displayed by the browser.
- Scroll down to the "Isolation source" text box to filter the data by source of isolation.
- Now the "Isolation source" filter box pops up. By default the top 100 unique values are shown, which can be viewed using the scrollbar. The number of items for each value are also shown. This box has a search bar to search for any values not displayed. Values can be selected and will update the number of items displayed in the table below. If two or more filters are open, then the selections in one filter will update the available values and unique items in the other filter. The filters that you see are generated on the fly to reflect the attributes of the isolates that you are currently viewing in the browser.
Identify the possible source of an outbreak
- Analyze data that's already available in the pathogen detection project by using the SNP Tree Viewer to view the phylogenetic relationships among a group of sequence-similar isolates from clincal or environmental sources.
For example, the FDA's GenomeTrakr project (BioProject PRJNA230969) for the surveillance and rapid detection of foodborne contamination events include a subset of E. coli isolates that belong to the SNP cluster "PDS000003441." Many of the isolates in that cluster were from an outbreak that originated in all-purpose flour. (Read more on the CDC website about that outbreak.)- or -
In the Isolates Browser display, you can click on the "PDS*" accession number that appears in the "SNP Cluster" column for any one of those isolates to open a Tree Viewer display for the SNP cluster and interactively examine the phylogenetic distance tree. A SNP cluster contains isolate genomes that have been found, via the Pathogens data processing pipeline, to be closely related.
The Tree View for SNP cluster PDS000003441 shows a number of clinical and environmental samples that are very closely related, in some cases, with a distance of zero SNPs between the clinical and environmental samples. (Mouse over any branch in the tree to view the SNP distance between the isolates.) The phylogenetic distance tree therefore sheds light on the possible source of the outbreak.
The sequence data analysis and SNP Tree Viewer help sections of this document provide additional details about SNP clusters and using the SNP Tree Viewer, respectively. The SNP Tree Viewer help includes an illustrated example of SNP Tree Viewer launch points and illustrated example of a SNP Tree Viewer display.
- Submit sequence reads to NCBI and obtain data analysis results on the Pathogen Detection project FTP site, in the form of phylogenetic distance trees that show the relationship of your isolates to those already in the Pathogen Detection project.
More examples...
- The Advanced Search > Examples of SOLR queries section of this document provides additional examples of field-specific searches and complex Boolean searches using the SOLR query syntax.
- The Pathogen Detection Reference Gene Catalog > Use cases/sample searches section of this document shows how to find antimicrobial resistance (AMR) genes, point mutations that confer resistance, AMR content of known isolates, and more.
Pathogens Project Components
Resources/Tools
Isolates Browser
The Isolates Browser was built to answer two specific questions for incoming pathogen genomes:
1) is this isolate clonally related to anything else in the database?
2) what is the AMR repertoire of this isolate?
It allows users to browse and search over 300,000 pathogen isolates, effectively and efficiently providing access to the National Database of Antibiotic Resistant Organisms.
Upon opening the Isolates Browser, a table displays data for all available isolates, with the most recently added data at the top. You can query the Isolates Browser with a wide variety of allowable search terms. The data can be sorted by clicking on column headers, filtered by using the "Filters" interface (e.g., Property: has antimicrobial resistance (AMR) genotypes), or searched using basic or advanced queries.
Every row in the Isolates Browser is an assembled isolate, possibly with antimicrobial resistance (AMR), virulence, and/or stress response genotype data, and antibiotic susceptibility (AST) phenotype data, as available.
If an isolate has a "PDS*" accession number in the "SNP Cluster" column, that indicates it is part of a SNP cluster. You can click on the PSD* accession to launch the SNP Tree Viewer and examine the relationships among your isolate of interest and other isolates that have been found, via the Pathogens data processing pipeline, to be closely related.
A separate section of this file provides Isolates Browser help documentation, with details on how the browser can be used, including allowable input, a decription of the output, and an illustrated example of search results.
SNP Tree Viewer
The SNP Tree Viewer displays a phylogenetic tree of pathogen isolates, built from assembled genomes by the maximum compatibility method. It shows relationships among the isolates based on the number of single nucleotide polymorphisms (SNPs) they contain relative to each other. Each tree represents a cluster of isolates that have been found, via the Pathogens data processing pipeline, to be closely related.
The trees can be used to examine the relationships of isolates in a SNP cluster to each other, and to identify the possible source of an outbreak based on the sequence similarity of the clinical and environmental isolates in a tree. (See an example in How to identify the possible source of an outbreak.)
A separate section of this file provides SNP Tree Viewer help documentation, with details on how the tree viewer can be used. It includes an illustrated example of SNP Tree Viewer launch points and illustrated example of a SNP Tree Viewer display.
Automatic e-mail notifications of new data
There are two ways to receive automatic e-mail notifications of new data, and you must be logged into your free My NCBI account to use either one:
"Save" a search in the Isolates Browser
- A "Save" button in the Isolates Browser interface allows you to save one or more searches, and automatically notifies you about new isolates that match the criteria of each saved search. (Read more and view an illustrated example.)
- A "Watch" button in the SNP Tree Viewer interface allows you to watch one or more selected isolates in a tree, and automatically notifies you about new isolates that fall within the SNP distance that you have specified from the watched isolate(s). (Read more and view an illustrated example.)
Antimicrobial Resistance (AMR) resources
As antimicrobial resistance (AMR) continues to evolve in many bacterial pathogens, the NCBI Pathogen Detection Project has developed a database to collect curated information about AMR genes, as well as tools to access the data. The AMR resources include:
- AMR Landing page - provides information about the NCBI National Database of Antibiotic Resistant Organisms (NDARO), a collaborative, cross-agency, centralized hub for researchers to access AMR data to facilitate real-time surveillance of pathogenic organisms.
- AMR Resources page - provides a list of available resources, with a brief description and sample searches or links to additional information about each one.
- Pathogen Detection Reference Gene Catalog
is a component of the Pathogens Isolates Browser. The Reference Gene Catalog is a non-redundant database of bacterial genes related to antimicrobial resistance, stress resistance, virulence, and antigenicity. A graphical user interface (GUI) allows you to browse and search the Catalog, which includes two data subsets:
- "Core": this subset includes highly curated, AMR-specific genes and proteins from the Bacterial Antimicrobial Resistance Reference Gene Database (BioProject PRJNA313047), plus point mutations. The sources of input for this curated database include: 1) allele assignments, 2) exchanges with other external curated resources, 3) reports of novel antimicrobial resistance proteins in the literature.
- "Plus": this subset includes genes related to biocide and stress resistance, general efflux, virulence, or antigenicity.
(The Pathogen Detection Reference Gene Catalog supercedes the previously available "AMR Reference Gene Browser," which encompassed only the "core" data set.) - AMRFinderPlus - a tool that compares isolate genomes against the reference protein set using BLAST and against the HMM set using HMMER, and uses the gene hierarchy to provide the most specific protein assignment to antimicrobial resistant protein or family, if present in the query set of proteins. The original AMRFinder identifies acquired antimicrobial resistance (AMR) genes, as well as point mutations that confer antimicrobial resistance, in either protein datasets or nucleotide data, including genomic data. AMRFinderPlus identifies the AMR genes and point mutations that are found by the original AMRFinder, plus it identifies select members of additional classes of genes such as virulence factors, biocide, heat, acid, and metal resistance genes. Unlike other AMR gene detection methods that report the best hit, AMRFinderPlus reports the specific gene symbol based on the available evidence. For example, when presented with a novel blaKPC allele that is nearly identical to blaKPC-2, closest hit tools might return blaKPC-2, but AMRFinderPlus would call it as blaKPC so that users do not incorrectly assume the phenotype (illustrated example). More details about the tool are provided in a publication by Feldgarden M, et al., 2019.
- Separate sections of this document provide more details about Antimicrobial Resistance (AMR) Resources and more details about AMRFinderPlus, including an illustration of the blaKPC example, as well as links to install AMRFinderPlus software, download data files, interpret AMRFinderPlus results, and read more. See AMRFinderPlus references for related publications
- MicroBIGG-E is the Microbial Browser for Identification of Genetic and Genomic Elements. Every row in the MicroBIGG-E display is an anti-microbial resistance (AMR), stress response, and/or virulence gene that has been identified in an isolate by the data processing pipeline, with information about the method used to identify it, supporting evidence, and the element's type, subtype, class, subclass, and more. The purpose of MicroBIGG-E is to enable researchers to obtain the actual contigs that contain a genetic or genomic element of interest, in order to conduct further analysis. The MicroBIGG-E help section provides information on how to use the tool.
- Submit sequence and phenotype data related to AMR - includes links to instructions how to: submit data for real-time analysis; submit antibiograms to the BioSample database; and request new alleles for Beta-Lactamase, MCR, and Qnr Genes.
- FTP/Raw Data Download - includes AMRFinderPlus data files and the Bacterial Antimicrobial Resistance Reference Gene Database (BioProject PRJNA313047)
- AMR resources schematic illustration - A separate section of this document provides more details about Antimicrobial Resistance (AMR) Resources, including a schematic illustration of AMR resources that shows the data sets and tools and the relationships among them. An antimicrobial resistance factsheet is also available on the FTP site.
FTP site
A separate section of this document provides an overview of the data available on the FTP site, and the FTP readme file provides additional details.
Data submission tools
A separate section of this document provides an overview of the data submission process, and links to detailed submission instructions.
Types of Data
BioProject records | BioSample records | Raw data: Sequence reads | Genomes | Genotypes: antimicrobial resistance (AMR) genes | Phenotypes: antimicrobial susceptibility test (AST) data (antibiograms)
BioProject records
- A BioProject is a collection of biological data related to a single initiative, originating from a single organization or from a consortium. A BioProject record provides users a single place to find links to the diverse data types generated for that project. As the sequence data archives (GenBank and SRA) require submission to a BioProject for assembled genomes, this means that every isolate in the Isolate Browser comes from one of these BioProjects. There may be many isolates from any particular BioProject.
- Example: Retrieve the BioProject PRJNA230969, which describes the GenomeTrakr project by the US Food and Drug Administration (FDA) to sequence Escherichia coli (E. coli) genomes for the surveillance and rapid detection of foodborne contamination events.
- Submit: See the data submissions section of this document for instructions on submitting BioProjects.
BioSample records
- BioSample records describe the biological source materials used in experimental assays. For many pathogen samples, a template/package is used that has a minimal set of required fields that was developed specifically for this project: (clinical package, environmental package).
- Example: Retrieve an individual BioSample record, SAMN05245394, for Escherichia coli isolated from all-purpose flour and sequenced as part of the FDA's GenomeTrakr project (BioProject PRJNA230969) for the surveillance and rapid detection of foodborne contamination events.
- Example: Retrieve all biosamples that are part of the FDA's GenomeTrakr project (BioProject PRJNA230969) for the surveillance and rapid detection of foodborne contamination events.
- Submit: See the data submissions section of this document for instructions on submitting BioSamples.
Raw data: Sequence reads
- Sequence Read Archive (SRA) stores raw sequencing data and alignment information from high-throughput sequencing platforms. Most of the major pathogen surveillance efforts use next generation sequencing platforms with raw sequence data deposited in SRA. The majority of isolates in the Isolate Browser have been assembled using the Pathogen Detection data processing pipeline from the raw data in SRA.
- Submit: See the data submissions section of this document for instructions on submitting sequence reads.
Genomes
- Pathogen genomes are from two sources: 1) assemblies submitted to the GenBank nucleotide sequence database from outside contributors, 2) genomes assembled in the Pathogen Detection data processing pipeline using the raw sequencing data in SRA. Currently NCBI is working on depositing these assemblies into GenBank, however the vast majority are not yet available there.
- Submit: See the data submissions section of this document for instructions on submitting assembled genomes.
- Note: Each Pathogen Detection Target ("PDT" record) in the Pathogen Detection Project contains the genome assembly for a single pathogen isolate.
- There are several types of genome assemblies in the Project:
- isolates submitted directly to GenBank as assembled genomes, and therefore have a corresponding "GCA" accession
- isolate genomes assembled by the NCBI Pathogens data processing pipeline from sequence reads, but not published as genome sequence records in GenBank
- isolate genomes assembled by the NCBI data processing pipeline and then submitted to GenBank either by the submitter or on behalf of the submitter with their permission.
Genotypes
- Antimicrobial resistance (AMR), virulence, and stress resistance genotypes are available in the Isolates Browser and are derived from the annotation of the assembled pathogen isolate using the NCBI AMRFinderPlus tool. (The data processing pipeline section of this document provides additional details about genome annotation.) For more information on NCBI antimicrobial resistance resources see this page.
- Search tip: To retrieve all pathogen isolates that have AMR genotype data, open the Pathogens Isolates Browser, click on the "Filters" menu, scroll down to the "Property" filter, and select the checkbox for the desired property, such as "has AMR genotypes," "has stress genotypes," and/or "has virulence genotypes". (See Isolates Browser help for more information about Filters, as well as information about how to search the "AMR Genotypes" data field directly.)
- Genotype categories: The genes that have been identified in an isolate's genome sequence are grouped into genotype categories, such as complete, partial, partial end of contig. The genotype categories appear when you use the Isolates Browser's choose columns function to display the genotype data columns. The AMR genotypes column is displayed by default, and you can use the choose columns function to display additional data columns, such as Stress genotypes and/or Virulence genotypes. (The data processing pipeline section of this document provides more information about genotype categories.)
- Additional details: A separate section of this document provides an overview of the Antimicrobial Resistance (AMR) resources that are available from the Pathogen Detection Project.
- Submit: See the data submissions section of this document for instructions on submitting antimicrobial resistance genes.
Phenotypes
- Antimicrobial Susceptibility Test (AST) data, also referred to as AST phenotypes or antibiograms, are included by submitters as data in BioSample records, when available. Those BioSample records with AST data can be retrieved from the BioSample database directly. For those BioSample records for which sequencing data is submitted, and which are also incorporated into the Pathogen resources, the Isolate Browser displays the antibiotic compounds from each antibiogram, binned into the SIR (sensitive, intermediate, resistance) calls as made by the submitter into a separate column: AST_phenotypes. You can submit AST data for your samples. See How to submit for information on how to submit that data
- Example: View BioSample SAMN05170351, an Escherichia coli isolate that was sequenced as part of a Multi-Site Gram-Negative Bacilli Surveillance Initiative by the CDC's Emerging Infections Program (EIP), BioProject PRJNA288601. The BioSample record includes antibiogram data for more than 20 antibiotics.
- Example: Retrieve all BioSample records that include antibiograms and that are from Escherichia coli.
- Search tip: To retrieve all pathogen isolates that have AST phenotype data, open the Pathogens Isolates Browser, click on the "Filters" menu, scroll down to the "Property" filter, and select the checkbox for "has AST phenotypes." (See Isolates Browser help for more information about Filters, as well as information about how to search the "AST Phenotypes" data field directly.)
Note that Isolates Browser default display does not show the AST_Phenotypes data column, but you can easily add it to the display by using the Choose columns option. - Submit: See the data submissions section of this document for instructions on submitting antimicrobial susceptibility test data (antibiograms).
- A list of possible phenotype values is shown on the BioSample Beta-Lactamase Antibiograms page, under the "Resistance Phenotype" tab, and includes:
- intermediate (I)
- nonsusceptible (NS)
- not defined (N, ND)
- resistant (R)
- susceptible (S, sensitive)
- susceptible-dose dependent (SSD)
Contributors
List of contributors by organism
- A list of major contributors by organism is accessible from the Pathogen Detection project home page.
The list is a set of contributors flagged by the NCBI Pathogen Detection team as of special interest and includes US Federal Government Agencies, State Health Departments and Agricultural Departments, Hospitals and Universities, and International Institutions. It DOES NOT represent every single sequence submitted to the International Nucleotide Sequence Database Collaboration (INSDC) OR the Sequence Read Archive (SRA) and incorporated into Pathogen Detection, as the list would be enormous. If you are interested in having your already submitted data tabulated here, then contact the NCBI Pathogen team at pd-help@ncbi.nlm.nih.gov.
Additional contributors
- The Pathogen Detection project continues to grow and welcomes data submissions from additional contributors. The data submissions section of this document provides an overview of the submissions process and links to pages that provide detailed instructions.
Data Retrieval & Analysis
- Text term searches
- Sequence data analysis
- Automatic e-mail notifications of new data
- Pathogen Detection Resources at Google Cloud Platform
Text term searches
Input text term(s)
- If you want to retrieve isolates from the existing data in the Pathogen Detection project, you can use the Isolates Browser to search for isolates that contain a term(s) of interest, as shown in the example below.
Example of text term search:
- Retrieve the set of isolates that contain the phrase "all-purpose flour".
The Isolates Browser Help section of this document provides information about allowable search terms, query tips about special characters such as hyphens in search terms, the use of quotes for phrase searches, and case sensitive vs. case insensitive searches, available data fields, and more.
Output tabular list of isolates that contain your search term(s)
- The Isolates Browser will display a table listing the isolates that contain at least one of the specified search terms (if you do a basic search), or the isolates that meet more stringent search criteria (if you do an advanced search).
Read more about the Isolates Browser output and see an illustrated example of search results.
More information about text term searches: tips and techniques
- The Isolates Browser Help section of this document provides additional information about basic searches, advanced searches using the SOLR query language, available data fields, examples of SOLR queries, and more.
Sequence data analysis
Real time analysis
- Unlike other NCBI system such as BLAST, the Pathogen Detection project is not built with an interactive interface that allows users to upload their data and immediately obtain an answer. Instead, this project was set up to facilitate interactive analyses of large-scale surveillance projects that are automatically submitting real-time data to the NCBI archives that are then routed to an automated pipeline that generates interactive web reports on a daily basis. The web displays allow users to search, browse, and filter the automatically analyzed data that has been already submitted.
Input sequence data
- If you have sequenced new isolates and want to determine their relationship to existing isolates in the Pathogen Detection project, then you can follow the data submission procedures described in a separate section of this document. Your submission(s) will go through the NCBI data processing pipeline, which includes sequence analysis to identify closely related isolates. The results of the analysis on your data are then made available on the FTP site and in the SNP Tree Viewer, as described in the example below.
- All of the existing isolates in the Pathogen Detection project have also undergone sequence analysis after they were submitted, and their results are also available on the FTP site and in the SNP Tree Viewer.
Output phylogenetic distance trees
- Sequence data analysis results in SNP Tree Viewer -- When you are viewing pathogens in the Isolates Browser, any isolate that belongs to a SNP cluster has a "PDS*" accession number in the "SNP Cluster" column of the Isolates Browser search results. The "PDS*" accession links to the SNP Tree Viewer, which displays an interactive phylogenetic tree of all the isolates in the SNP cluster.
(See the SNP Tree Viewer Help section of this document for additional details on how to use that tool. It includes an illustrated example of SNP Tree Viewer launch points and illustrated example of a SNP Tree Viewer display.) - Sequence data analysis results on FTP -- The phylogenetic distance trees are also available in the Pathogen FTP site, under the "Results" directory. (A separate section of this document provides more information about the FTP site.)
Example of sequence data analysis results (as interactive displays in SNP Tree Viewer)
- The FDA's GenomeTrakr project (BioProject PRJNA230969) for the surveillance and rapid detection of foodborne contamination events include a subset of E. coli isolates that belong to the SNP cluster "PDS000003441." Upon submission to NCBI, those isolates were compared to all other isolates in the Pathogen Detection project and were found, via the Pathogens data processing pipeline, to be closely related to other isolate genome sequences in that SNP cluster. In the Isolates Browser display, you can click on the "PDS*" accession number that appears in the "SNP Cluster" column for any one of those isolates (e.g., isolate PDT000133982.1) to open a SNP Tree Viewer display for the SNP cluster and interactively examine the phylogenetic distance tree. The Tree View shows a number of clinical and environmental samples that are very closely related, and therefore sheds light on the possible source of the outbreak. (Read more on the CDC website about that outbreak.)
More information about pathogen sequence data analysis
- A separate section of this document provides more information about the SNP Tree Viewer.
- A separate section of this document provides more information about the FTP site.
Automatic E-mail Notifications of New Data
There are two ways to receive automatic e-mail notifications of new data, and you must be logged into your free My NCBI account to use either one:
"Save" a search in the Isolates Browser
- A "Save" button in the Isolates Browser interface allows you to save one or more searches, and automatically notifies you about new isolates that match the criteria of each saved search. (Read more and view an illustrated example.)
"Watch" an isolate of interest in the SNP Tree Viewer
- A "Watch" button in the SNP Tree Viewer interface allows you to watch one or more selected isolates in a tree, and automatically notifies you about new isolates that fall within the SNP distance that you have specified from the watched isolate(s). (Read more and view an illustrated example.)
Isolates Browser help
- What is the Isolates Browser?
- Input to Isolates Browser
- Allowable search terms
- Basic Search
- Advanced Search: SOLR Query Language
- Query terms
- Operators
- Parentheses
- Data fields
- AMR Genotypes (AMR_genotypes)
- AMR Genotypes core (AMR_genotypes_core)
- AST Phenotypes (AST_phenotypes)
- Collection Date (collection_date)
- Create Date (creation_date)
- Food origin (food_origin)
- Isolation Source (isolation_source)
- Isolation type (epi_type)
- Location (geo_loc_name)
- Minimum SNP distance within same isolation source type (minsame)
- Minimum SNP distance across different isolation source types (mindiff)
- Organism Group (taxgroup_name)
- Strain (Strain)
- Serovar (serovar)
- see a list of all Isolates Browser data fields...
- Search modifiers
- Examples of SOLR queries
- Isolation source:
Show all isolates that have the exact phrase "All-Purpose Flour" in the isolation_source data field - Geographic location and organism group:
Show all Salmonella isolates collected in USA - Genotypes: isolates that have specific genes:
Show all of the isolates that have a mobile colistin resistance gene and a KPC beta-lactamase - Phenotypes: isolates that are resistant to a given antibiotic:
Show all of the isolates that are resistant to ciprofloxacin - Genotypes and phenotypes:
Example 1: missing specific genes, resistant to antiobiotic:
Show all of the isolates that lack both a blaKPC and blaNDM carbapenemase but are resistant to imipenem
Example 2: has specific gene, susceptible to antiobiotic:
Show all of the isolates that have a blaKPC gene and are susceptible to meropenem
Example 3: has specific gene, resistant to either of two antiobiotics:
Show all of the isolates that have a qnr gene and that are resistant to either ciprofloxacin or nalidixic acid - Where is my isolate?
Retrieve your data by BioSample accession numbers or SRA Run accession numbers - Batch search with isolate identifiers that contain a hyphen:
Input a list of isolate identifiers that contain special characters (e.g., hyphens)
- Isolation source:
- Output from Isolates Browser
 Search/retrieve isolates browser data in Google Cloud BigQuery
Search/retrieve isolates browser data in Google Cloud BigQuery- Tabular list of isolates
- Exceptions table
- Matched clusters
- Filters to refine results
- Sort order
- Customize the Isolates Browser display
- SNP Tree Viewer link for each isolate that belongs to a SNP cluster
- "Share" function in the Isolates Browser
- Illustrated example of Isolates Browser search results
- "Save" function to receive automatic e-mail notifications about new data from a saved search
- Display isolates in MicroBIGG-E using Cross-browser selection
- Download data from the Isolates Browser web display
What is the Isolates Browser?
Upon opening the Isolates Browser, a table displays data for all available isolates, with the most recently added data at the top. The data can be faceted by using filters (e.g., Property: has antimicrobial resistance (AMR) genotypes), queried with a wide variety of allowable search terms, using either basic or advanced search methods, and sorted by clicking on column headers.
Every row in the Isolates Browser is an assembled isolate, possibly with antimicrobial resistance (AMR), virulence, and/or stress response genotype data, and antibiotic susceptibility (AST) phenotype data, as available.
The table summarizes and links to the data available for each pathogen, such as strain name, geographic origin, isolation type (environmental or clinical), BioSample UID, organism group (PDG* accession), antimicrobial resistance (AMR)/virulence/stress response genotypes, and antibiotic susceptibility (AST) phenotypes, and more (see list of data fields available in the Isolates Browser).
If an isolate has a "PDS*" accession number in the "SNP Cluster" column, that indicates it is part of a SNP cluster, and you can click on the PSD* accession to launch the SNP Tree Viewer and examine the relationships among your isolate of interest and other similar isolates.
The information below provides details on how the Isolates Browser can be used, including allowable input, a description of the output, and an illustrated example of search results. The browser accepts basic queries that contain one or more text terms, with or without quotes. It also accepts advanced queries using the SOLR query language, such as complex Boolean queries that look for the search terms in specific data fields. Filters on the results page enable you to further narrow your retrieval, if desired, and links from the Isolates Browser to the SNP Tree Viewer enable you to interactively explore the relationship of an isolate of interest to other isolates in the SNP cluster, which were found, via the Pathogens data processing pipeline, to have closely related genome sequences.
Input for Isolates Browser
Basic search | Query tips | multiple terms | special characters | phrase searches | advanced searches | case sensitive vs. case insensitive searches
Filters to refine search | filters menu options | filters are generated on the fly | look for synonyms within a filter
Advanced search | SOLR query language | Query terms | Operators | Parentheses | Data fields | Examples of SOLR queries
Allowable search terms
- The Isolates Browser accepts a variety of text strings as input. For example, the browser can retrieve isolates that contain terms such as lettuce or romaine or strawberry or strawberries or "all-purpose flour". You can also search for many other types of text strings, such as organism group, antimicrobial resistance (AMR) genotype, Antimicrobial Susceptibility Test (AST) phenotype, and more. The data fields section of this document lists the many types of strings that can be searched, and provides example searches for the various data fields.
Free text vs. controlled vocabulary
- Free Text - Many data fields in the Isolates Browser are free text and therefore contain the exact terms that were supplied by the data submitters.
- Please note that data submitters might use different forms of a term in their submissions.
- For example, some submitters might use hyphens between terms (e.g., "all-purpose flour") while others might use spaces (e.g., "all purpose flour").
- For a comprehensive search, include synonyms in your query, or use wildcards to search for a word stem.
- For a more precise search, you can limit your query to a specific data field, such as Isolation source. Please note that, in field-specified queries, both the data field names and values are case sensitive.
- Separate sections of this document provide query tips about searching for synonyms, and describe how the Isolates Browser handles special characters in search terms (such as hyphens in strain names, parentheses in gene names, slashes in serovar names, etc.).
- Controlled Vocabulary - Some data fields in the Isolates Browser contain a controlled vocabulary. In these fields, it is not necessary to search for synonyms.
- For example, the Location data field, which lists the geographic location where the sample was isolated, contains two parts: Country and Region. Country is a controlled vocabulary (https://www.ncbi.nlm.nih.gov/genbank/collab/country). Region is not controlled and can be anything (i.e., free text, such as a state abbreviation, province name, city name, zip code, etc.).
Unique identifiers and NCBI accession prefixes
- NCBI Unique identifiers, such as an NCBI accessions (e.g., biosample ID SAMN05245394, bioproject ID PRJNA230969, etc.) can be used to retrieve pathogen isolates.
Note that while NCBI accessions are unique, there can be multiplicity involved and it is possible for the same accession to appear in multiple current Pathogen records. For example, two or more isolates can belong to the same BioProject and/or same SNP cluster, so the record for each isolate will have its own PDT accession, but all of those records will contain the same PRJ and/or PDS accession. - Some NCBI accessions that can be searched in Pathogens Detection Project have the following prefixes:
GCA | GCF | NG | PDG | PDS | PDT | PRJ | SAMN | SRR | SRS | WP
- GCA_ - Accession number prefix for a GenBank genome assembly. This is data submitted by the scientific community directly to GenBank as an assembled genome.
(Read more about genomes in the data types section of this document.) - GCF_ - Accession number prefix for a RefSeq genome assembly. This is a representative genome assembly for a given organism in RefSeq, a non-redundant database.
(Read more about Prokaryotic RefSeq Genomes.)
(Read more about NCBI Genome Assembly Models.) - NG_ - Accession number prefix for a RefSeq genomic sequence record.
(Read more about NG_* accessions.) - PDG - Accession number prefix for a Pathogen Detection Organism Group.
Technical note: An organism group (PDG) contains one or more targets (PDTs). A PDT is a member of zero or one SNP cluster (PDS), and never more than one cluster. A SNP cluster is composed of two or more PDTs, and each ach PDS is completely contained within a PDG.
(Read more about organism groups in the data fields section of this document.) - PDS - Accession number prefix for a Pathogen Detection SNP Cluster.
(Read more about SNP clusters in the data fields section of this document.) - PDT - Accession number prefix for a Pathogen Detection Target. This is the Pathogen project accession for an individual isolate's genome assembly.
(Read more about genome asemblies in the data types section of this document.) - PRJ - Accession number prefix for an International Nucleotide Sequence Database Collaboration (INSDC) BioProject.
(Read more about bioprojects in the data types section of this document.) - SAMN - Accession number prefix for a NCBI BioSample.
(EBI BioSamples have the prefix SAMEA, and DDBJ BioSamples have the prefix SAMD.)
(Read more about biosamples in the data types section of this document.) - SRR - Accession number prefix for a NCBI Sequence Read Archive (SRA) Run. A Run is an object that contains actual sequencing data for a particular sequencing experiment. SRA experiments may contain many Runs depending on the number of sequencing instrument runs that were needed.
(Read more about SRA accessions.) - SRS - Accession number prefix for a NCBI Sequence Read Archive (SRA) Experiment Sample. A Sample is an object that contains the metadata describing the physical sample upon which a sequencing experiment was performed. That information is imported from the BioSample record.
(Read more about SRA accessions.) - WP_ - Accession number prefix for a RefSeq protein sequence that has been found in one or more archaeal and bacterial RefSeq genomes. If the identical protein sequence has been found in multiple genomes, the WP_ sequence record is a non-redundant representation of all the instances of the protein, and includes links to the genomic sequences that code for the protein.
Details about WP_* accessions are provided on the web pages that describe the RefSeq non-redundant proteins, the Prokaryotic RefSeq Genome Re-annotation Project, and the New RefSeq protein product and data model.
- GCA_ - Accession number prefix for a GenBank genome assembly. This is data submitted by the scientific community directly to GenBank as an assembled genome.
Basic Search
Query tips | multiple terms | special characters | phrase searches | advanced searches | case sensitive vs. case insensitive searches
Filters to refine search | filters menu options | filters are generated on the fly | Filters for gene fields | Filter for Scientific name | look for synonyms within a filter
- Query tips
multiple terms | special characters | phrase searches | advanced searches | case sensitive vs. case insensitive searches
- Multiple search terms
If you enter multiple search terms, the system will automatically separate the terms with a Boolean OR, and will therefore retrieve isolates that have at least one of the terms in your query. Note, that this is different than many other NCBI resources that require explicity use of "OR" in the search. For isolates that have more than one term from your query, please note that the terms will not necessarily be adjacent to each other, and will not have to be present in the same data field. (If desired, use quotes to force a phrase search, and use an advanced search to limit searches to a specific data field.)
For example, try a search for the following query (with no quotes and no special characters):
all purpose flour
The search system interprets the query as three separate terms:
all OR purpose OR flour
It therefore retrieves isolates whose records contain the term "all" in any data field, or the term "purpose" in any data field, or the term "flour" in any data field.
- Special characters
If a search term contains special characters (such as hyphens in strain names, parentheses in gene names, slashes in serovar names, etc.), the system recognizes the special characters as part of the search term, and only retrieves isolates that include the term exactly as it was entered.
For example, try a search for the following query (with no quotes but with a hypen):
all-purpose flour
The search system treats the hyphen as part of the search term and interprets the query as two separate terms:
all-purpose OR flour
It therefore retrieves isolates whose records contain the hyphenated term "all-purpose" in any data field, or the term "flour" in any data field.
A separate section of this document provides additional information about special characters.
- Phrase searches using quotes
Use quotes around a set of words if you want to search all of the words together as a phrase. That will retrieve isolates which have the terms adjacent to each other, and the phrase can occur in any data field of the record. (If desired, use an advanced search to limit searches to a specific data field.)
For example, try a search, with a hyphen and with quotes, for:
"all-purpose flour".
The quotes require the search system to interpret the query as the complete phrase:
all-purpose flour
It therefore only retrieve isolates whose records contain that exact character string.
If no quotes are used, the system will automatically insert a Boolean OR when it encounters a space in the query string. In the example above, if the quotes were absent, the system would interpret the search as all-purpose OR flour. The hypen would be retained because the search system recognizes special characters as part of the search term.
- Advanced searches
Use an advanced search strategy if you want to exercise more control over the search, such as specify the desired Boolean operators; use parentheses to specify a list of items or to determine the order of execution of the query; and/or limit your search to a specific data field. For queries that specify data fields, please note that both the data field names and values are case sensitive.
Examples of advanced searches are available in a separate section of this document and include:
- Isolation source:
Show all isolates that have the exact phrase "All-Purpose Flour" in the isolation_source data field - Geographic location and organism group:
Show all Salmonella isolates collected in USA - Genotypes: isolates that have specific genes:
Show all of the isolates that have a mobile colistin resistance gene and a KPC beta-lactamase - Genotypes and phenotypes:
Example 1: missing specific genes, resistant to antiobiotic:
Show all of the isolates that lack both a blaKPC and blaNDM carbapenemase but are resistant to imipenem
Example 2: has specific gene, susceptible to antiobiotic:
Show all of the isolates that have a blaKPC gene and are susceptible to meropenem
Example 3: has specific gene, resistant to either of two antiobiotics:
Show all of the isolates that have a qnr gene and that are resistant to either ciprofloxacin or nalidixic acid - Where is my isolate?
Retrieve your data by BioSample accession numbers or SRA Run accession numbers - Batch search with isolate identifiers that contain a hyphen:
Input a list of isolate identifiers that contain special characters (e.g., hyphens)
- Isolation source:
-
Case sensitive versus case insensitive searches:
Case sensitive searches:
If you want to do an advanced search in order to query specific data fields, please note that the names of data fields, and the values they contain, are case sensitive.
The values in the data fields represent text strings exactly as they were entered by the submitter, including upper case and lower case letters, special characters such as hyphens, etc.
The case-sensitivity and the retention of special characters such as hyphens and parentheses (when they are internal to a search term) were built into the system in order to ensure precise handling of searches for values such as strain name, serovar, gene symbol, and more. The case sensitivity and handling of special characters applies to other data fields as well.
Therefore, when you search a specific data field, the system will retrieve isolates that contain the exact string you have specified, including upper case and lower case letters, as well as special characters such as hyphens and parentheses.
For example, search the Isolation Source data field for the phrase "All-Purpose Flour" by entering the following query:
isolation_source:"All-Purpose Flour"
The system will retrieve isolates that contain the exact string you have specified, including upper and lower case and the hyphen.
Case insensitive searches:
If you are uncertain about whether to use upper case or lower case letters in your query, then simply enter your search terms, regardless of case, without a data field specifier.
For example, any one of the following three queries will work equally well:
"all-purpose flour"
-- or --
"ALL-PURPOSE FLOUR"
-- or --
"All-Purpose Flour"
Those queries work because, in the absence of a data field specifier, the system searches a text index, which is a case insensitive compilation of terms from many text-containing data fields. Such searches are less precise in their retrieval, because the query terms can appear in any text field of pathogen isolate records; however, they provide a flexible data retrieval mechanism.
(Note: The presence or absence of the hyphen *will* affect the results, whether or not you specify a data field. This is because the hyphen is considered to be part of the search term and therefore must be present in the retrieved isolates. A separate section of this document provides additional information about special characters, as well as the use of quotes to force a phrase search.)
- Multiple search terms
Filters to refine search
- The "Filters" menu options
The "Filters" menu options in the Isolates Browser enable you to facet or subset the data in a variety of ways, and therefore can be used to refine your results, whether you have done a basic search or an advanced search.
. The filter menu now allows all data fields in the column chooser to be filtered. By default, each filter displays the top 100 terms (based on the number of isolates retrieved by a term). Note that:- A Boolean "OR" is applied if multiple items are checked in the same filter field. This way you can choose multiple values in the same filter. For example:
- Open the "Filters" tab of the Isolates Browser, scroll to the "Isolation source" field, and check the boxes for "stool" and "feces" The system will retrieve isolates that have either one of those values in the "Isolation source" field.
- A Boolean "AND" is applied if you select items in several different filter fields (Location, Source, etc). For example:
- Open the "Filters" tab of the Isolates Browser, then check the boxes for "clinical" in the "Isolation type" filter and "wound" in the "Isolation source" filter. The system will retrieve isolates that have both of your specified criteria.
- If you prefer to apply a Boolean "AND" to multiple items within the same filter field, you can enter a SOLR query. For example:
- To retrieve isolates that have both AMR genotypes AND Stress genotypes, open the Isolates Browser and enter a SOLR query such as:
property:"has AMR genotypes" AND property:"has stress genotypes".
(A separate section of this document provides additonal information about the SOLR query language. Additionally, note that the default columns in the Isolates Browser output include AMR genotypes but not Stress genotypes; use the choose columns function in the Isolates Browesr to add additional columns to the display.)
- To retrieve isolates that have both AMR genotypes AND Stress genotypes, open the Isolates Browser and enter a SOLR query such as:
property:"has AMR genotypes" AND property:"has stress genotypes".
- A Boolean "OR" is applied if multiple items are checked in the same filter field. This way you can choose multiple values in the same filter. For example:
- Filters are generated on the fly for a given data set
The choices listed in the "Filters" tab reflect the attributes of the isolates that you are currently viewing in the browser. By default the top 100 terms (based on the number of isolates retrieved by a term, and listed by count of isolates per value are displayed). The total number of unique values is also shown at the bottom of each filter tab. - Searching within filters
A search box is available in each filter to search for values not found in the top 100. Example, open the "Isolation source" filter and type in the word "chicken" in the search box. All matches to any of the values of the word "chicken" will show up, which is very useful to catch those synonymous values from multiple submitters.
Numeric fields have ranges that can be selected using the check button or reset, while date fields have a range selection as well as quick access to common recent queries.
. The search box can be reset with the reset button beside the search box. The entire filter can be removed with the 'X' at the top right corner.
Filters can be collapsed if more than one is shown with the double left hand arrow at the bottom left, and opened again after collapse with the double right hand arrow on collapsed tabs. Each tab is labeled with the filter name.
- Gene fields: AMR genotypes, Stress genotypes, Virulence genotypes, AMR genotypes core
The fields with gene and point-mutation fields have filters that separate the genes into categories based on characteristics that help to determine how likely the gene/point-mutation is to be properly transcribed and assembled. They are divided into COMPLETE, POINT, PARTIAL, HMM, MISTRANSLATION, and PARTIAL_END_OF_CONTIG. More information on what the categories mean is available below and on the AMRFinderPlus wiki. Each of the categories can be expanded by clicking on the '+' sign next to it, and within that the gene symbols may be selected to further refine your search. As with the other filter fields only the 100 most-frequent gene symbols will appear in the filter box. To search for specific genes you can use the search function within the filter.
- Scientific name
The Scientific name field is set up with a hierarchy that represents lineages based on NCBI Taxonomy to allow you to filter for all members of a given taxonomic group. Clicking on a node at a higher level will select all the taxa within that group even though the boxes by those names won't show up as selected. As with other filters only the 100 most common values are shown, in this case that is the 100 most common values in the Scientific name field and the higher level taxa that they belong to. You can search within the values using the Search box within the filter to narrow the choices and reveal scientific names that are not in the most frequent set. For example selecting Enterobacteriaceae will include all isolates that are Salmonella, E. coli, Shigella, and Klebsiella even though those more specific taxa are not selected individually.
For example selecting Enterobacteriaceae will include all isolates that are Salmonella, E. coli, Shigella, and Klebsiella even though those more specific taxa are not selected individually.
- Look for synonyms
A number of data fields do not use a controlled vocabulary, but instead list the various terms that submitters applied to their data. As a result, submitters might use different terms for the same concept. Therefore, if you are using filters, look for synonymous terms that are listed under a given filter and check the boxes for any/all terms that are of interest. If you are searching the data fields directly (as described in the advanced search section of this document), consider including synonyms in your query in order to broaden retrieval.
Synonyms are also useful to include if you are doing advanced searches, such as limiting your search to specific data fields. As an example, see the sample searches of the host organism data field.
Advanced Search
SOLR Query Language
The Isolates Browser uses a modified SOLR search platform (version 6.6) to retrieve pathogen data. The Apache SOLR Reference Guides provides detailed documentation for the platform. Some key concepts are introduced below, and link to the complete documentation in the SOLR Reference Guide 6.6, particularly the sections on: Searching > Query Syntax and Parsing > The Standard Query Parser.In some instances, there might be some slight differences between the Isolates Browser and the SOLR Standard Query Parser. For example, the Isolates Browser has been modified relative to the SOLR Standard Query Parser in the way it handles special characters that are part of a search term. Specifically, the browser has been programmed to automatically escape special characters (such as hyphens in strain names, parentheses in gene names, slashes in serovar names, etc.) and to treat them as part of the search term. The Browser therefore retrieves isolates that include the term exactly as it was entered, including special characters.
Query terms | single term | multiple terms | phrase | term modifiers | wildcard searches | special characters in search terms
Operators | AND, OR, NOT | plus (+) and minus (-) symbols | range searches [nnnn TO nnnn]
Parentheses | use to order Boolean queries | use to input a list of query terms | automatically escaped if part of a gene name or query term
Data Fields
Search modifiers
Examples of SOLR queries
SOLR Query terms
- Single term
A single query term, such as lettuce, will retrieve all isolates that have the term in any data field.
Examples:
A search for:
lettuce
will show isolates that contain the term lettuce in any data field.
Tips:
If you search a specific data field, your search will become case sensitive.
For example, compare the search results for:
isolation_source:lettuce
versus:
isolation_source:Lettuce
For broader retrieval, you can either remove the data field specifier to conduct a case insensitive search, or include synonyms in your query, for example:
isolation_source:lettuce OR isolation_source:Lettuce
A separate section of this document provides additional information about searching for synonyms.
-
Multiple terms
If you include two or more terms in your query, the system will automatically insert a Boolean OR in each space that it encounters. As a result, it will search for each word individually, and the system will show isolates that contain at least one of your search terms in any data field.
Examples:
A search for the following query (with no quotes or special characters):
romaine lettuce
will be interpreted as:
romaine OR lettuce
A search for the following query (with no quotes or special characters):
all purpose flour
will be interpreted as:
all OR purpose OR flour
because the system will insert an OR when it encounters each space in the query string.
A search for the following query (with no quotes and with a hyphen in all-purpose):
all-purpose flour
will be interpreted as:
all-purpose OR flour
because the system will treat the special character (hypen) as part of the first query term, and it will insert an OR where it encounters a space in the query string.
Tips:
If you include a data field specifier and you do not enclose your query terms in quotes, the data field specifier will be applied only to the term that immediately follows it, and that term will be searched in a case sensitive manner.
For example, a search for the following query (with no quotes or special characters):
isolation_source:romaine lettuce
will be interpreted as:
isolation_source:romaine OR lettuce
The system will show all isolates that have the lower case term romaine in the Isolation Source data field, and the term lettuce in any case and in any field.
If, on the other hand, you want to search romaine lettuce as a phrase, you will need to use quotes, as described below. -
Phrase
If you want to search for a phrase, surround your query terms with quotes.
Examples:
A search for:
"romaine lettuce"
will show isolates that contain that phrase in any data field.
A search for:
"all-purpose flour"
will show isolates that contain the phrase all-purpose flour, and will conduct the search in a case insensitive manner because the query does not include a data field specifier.
A search for:
isolation_source:"All-Purpose Flour"
and will show isolates that contain the phrase All-Purpose Flour in the Isolation Source data field.
Because the query includes a data field specifier, the search is conducted in a case sensitive manner. It will therefore only show isolates that have the exact phrase you specified, including upper and lower case letters as well as the hypen.
Tips:
If no quotes are used, the system will automatically insert a Boolean OR when it encounters a space in the query string. If you query specific data fields, please note that the names of data fields, and the values they contain, are case sensitive. Special characters, such as the hypen in the examples above, are recognized as part of the search term and therefore retained in the query, regardless of whether quotes are used.
For example, if the last sample search above was entered as isolation_source:All-Purpose Flour, with no quotes, it would be interpreted as isolation_source:All-Purpose OR flour. The Browser would show isolates that contain the term All-Purpose in the Isolation Source data field or the term flour in any data field. This is because the system processes the term adjacent to the data field specifier in a case sensitive manner, and inserts a Boolean OR when it encounters a space. -
Term modifiers
As noted in the "Standard Query Parser" section of the SOLR Reference Guide 6.6, "Solr supports a variety of term modifiers that add flexibility or precision, as needed, to searches. These modifiers include wildcard characters, characters for making a search "fuzzy" or more general, and so on." -
Wildcard searches
A question mark (?) can be included your query string to match any single character.
An asterisk (*) can be included your query string to match zero or more sequential characters.
Examples:
A search for:
AMR_genotypes:tet(*)
will show isolates that have a string of "tet(*)" in the AMR Genotypes data field, with the question mark serving as a wildcard to retrieve gene names that have any single character in the parentheses, such as tet(A), tet(M), tet(O), tet(X). etc.
A search for:
strawberr*
will show isolates that contain terms such as strawberry, strawberries, etc. in any data field.
A search for:
isolation_source:*berry
will show isolates that contain terms such as strawberry, mulberry, etc. in the Isolation Source data field.
Tips:
The wildcard characters can appear anywhere in your search term (at the beginning, middle, or end).
The SOLR Reference Guide 6.6 provides additional details about the use of wildcards.
-
Special characters in search terms
As noted in the introduction to the advanced search section of this document, the Isolates Browser uses the SOLR search platform (version 6.6) to retrieve pathogen data. However, in some instances, there might be some slight differences between the Isolates Browser and the SOLR Standard Query Parser.
For example, the Isolates Browser has been modified relative to the SOLR Standard Query Parser in the way it handles special characters that are part of a search term. Specifically, the browser has been programmed to automatically escape special characters (such as hyphens in strain names, parentheses in gene names, slashes in serovar names, etc.) and to treat them as part of the search term. As a result, the Browser retrieves isolates that include the term exactly as it was entered, including special characters.
Examples:
A search for:
strain:KCRI-598A
will show isolates that contain the term KCRI-598A in the Strain data field.
A search for:
serovar:1/2a
will show isolates that contain the term 1/2a in the Serovar data field.
A search for:
AMR_genotypes:ant(6)-Ia AND AMR_genotypes:aph(3')-IIIa
will show isolates that contain that have both the ant(6)-Ia and the aph(3')-IIIa in the AMR Genotypes data field.
Tips:
When you query specific data fields, please note that the names of data fields, and the values they contain, are case sensitive. Also, if your query string includes a space, surround the query string with quotes in order to do a phrase search. If no quotes are used, the system will automatically insert a Boolean OR when it encounters a space in the query string.
SOLR Operators
-
AND, OR, NOT
The SOLR search platform allows you to apply Boolean logic to queries with the AND, OR, and NOT operators. Boolean operators must be written in upper case letters, or they can be represented as symbols:
AND can be represented as &&
OR can be represented as ||
NOT can be represented as !
By default, the system applies the OR operator each time it encounters a space in the query string.
Examples:
A search for:
all-purpose flour
will be interpreted as:
all-purpose OR flour
because the system applies a Boolean OR when it encounters a space in your query string.
The system recognizes special characters such as the hyphen when they are part of a search term, and therefore will show isolates that contain the term all-purpose in any data field, or the term flour in any data field.
A search for:
romaine lettuce
will be interpreted as:
romaine OR lettuce
will show isolates that contain the term romaine in any data field, or the term lettuce in any data field.
A search for:
romaine AND lettuce
will show isolates that contain both of the terms, which can appear independently of each other in any data field. If you instead prefer to retrieve isolates in which two or more query terms to appear adjacent to each other, use quotes to conduct a phrase search. For example, a search for: "romaine lettuce" will retrieve isolates that contain that phrase romaine lettucein any data field.
A search for:
lettuce NOT romaine
will show isolates that contain the term lettuce, but not the term romaine.
That same search can also be written as:
lettuce !romaine
or as:
lettuce -romaine
Tips:
The SOLR Reference Guide 6.6 provides additional details about the use of Boolean operators. -
Plus (+) and Minus (-) symbols
The plus (+) and minus (-) symbols can be used to require that a term be present or absent, respectively, in the records retrieved by a search.
Examples:
A search for:
lettuce +romaine
will show isolates that contain the term lettuce (in any data field) and that must contain the term romaine (in any data field).
A search for:
lettuce -romaine
and will show isolates that contain the term lettuce (in any data field) but must not contain the term romaine (in any data field).
Tips:
The SOLR Reference Guide 6.6 provides additional details about the use of the plus (+) and minus (-) symbols in the section on Boolean operators. -
Range searches [nnnn TO nnnn]
To search for a range of values, enter a query such as:
collection_date:[value1 TO value2]
with square brackets surrounding the query string, and with the word "TO" written in upper case.
Examples:
Range of Collection Dates:
A search for:
collection_date:[2013-02* TO 2013-08*]
will show isolates that were collected anytime from February 2013 through August 2013.
A search for:
collection_date:[2013* TO 2015*]
will show isolates that were collected in any month or date from 2013 through 2015.
See the section of this help document for more information about the Collection Date data field, which accepts an asterisk (*) as a wildcard.
Range of Create Dates:
A search for:
creation_date:[2013-02 TO 2013-08]
will show isolates that were first seen by the Pathogen Detection system anytime from February 2013 through August 2013.
See the section of this help document for more information about the Create Date data field, which does NOT accept an asterisk (*) as a wildcard.
Tips:
The SOLR Reference Guide 6.6 provides additional details about Range searches.
Parentheses
-
Use parentheses to determine order of execution in Boolean queries
By default, the system applies Boolean operators from left to right in the query. Parentheses can be used to alter the order of execution of Boolean operators. Sub-queries that are surrounded by parentheses will be executed first.
Examples:
A search for:
AMR_genotypes:qnr* AND (AST_phenotypes:ciprofloxacin=R OR AST_phenotypes:"nalixidic acid=R")
will show all of the isolates that have a qnr gene and that are resistant to either ciprofloxacin or nalidixic acid.
(For additional information about this example, see the section of this help document on Examples of SOLR Queries > Genotypes and phenotypes: has specific gene, resistant to antiobiotics.)
Tips:
The SOLR Reference Guide 6.6 provides additional details about use of parentheses for grouping terms to form sub-queries. -
Use parentheses to input a list of query terms
Search terms that are enclosed in parentheses will be OR'ed together.
Examples:
A search for:
escherichia AND (FDA CDC USDA)
will show isolates that contain the term escherichia (in any data field), and the term FDA or CDC or USDA (in any data field).
-
Parentheses are automatically escaped if they are an internal part of a gene name or query term
As noted in the introduction to the advanced search section of this document, the Isolates Browser uses the SOLR search platform (version 6.6) to retrieve pathogen data. However, in some instances, there might be some slight differences between the Isolates Browser and the SOLR Standard Query Parser.
For example, the Isolates Browser has been modified relative to the SOLR Standard Query Parser in the way it handles special characters that are part of a search term. Specifically, the browser has been programmed to automatically escape special characters, such as parentheses that are part of gene names, and to treat them as part of the search term. As a result, the Browser retrieves isolates that include the term exactly as it was entered, including special characters.
Examples:
A search for:
AMR_genotypes:ant(6)-Ia AND AMR_genotypes:aph(3')-IIIa
will show isolates that contain that have both strings, ant(6)-Ia and aph(3')-IIIa, in the AMR Genotypes data field.
Data fields in the Pathogens Isolates Browser
Each data field reflects an available column in the Pathogens Isolates Browser web interface. The output section of this document provides tips on how to choose columns to include in the display.
Please note: in the list of available data fields below:
- The term shown in the regular font is the display name (column header) shown by the Isolates Browser web interface. The term shown in (italics) is the name of the corresponding data field, if you want to search that field directly.
- For example, one data field is listed as: Location (geo_loc_name). The term "Location" appears in the Isolates Browser column header, and "geo_loc_name" is the string you should use if you want to search that data field directly.
- Brief italicized search examples are also provided for each data field, when possible, showing how to query the data field directly. The values represent text strings exactly as they appear in the data fields, including upper case and lower case letters, special characters such as hyphens, etc. The data field names and values are case sensitive, as noted below.
The case-sensitivity and the retention of special characters such as hyphens and parentheses (when they are internal to a search term) were built into the system in order to ensure precise handling of searches for values such as strain name, serovar, gene symbol, and more. The case sensitivity and handling of special characters applies to other data fields as well.
Therefore, when you search a specific data field, the system will retrieve isolates that contain the exact string you have specified, including upper case and lower case letters, as well as special characters such as hyphens and parentheses.
Case insensitive searches: If you are uncertain about the exact text string that appears in isolate records, then you can simply enter the query in any text format (all upper, all lower, or mixed case) without a data field specifier. The system will then search the Text index, which is a case insensitive compilation of terms from many text-containing data fields. This provides a flexible search mechanism, although it is less precise in its retrieval as the query terms can appear in any text field of the pathogen isolate records. (A separate section of this document provides examples of case insensitive searches.)
The query tips section of this document includes a comparison of case sensitive versus case insensitive searches.
The available data fields in the Pathogens Isolates Browser include the following:
Note that fields shown in the default display are highlighted in blue. Each field is written in this format: Display name (data_field_name)
The "Display name" is the column header that appears in the Isolates Browser web interface, and the "data_field_name" is the case-sensitive string you should enter if you want to search the data field directly using a SOLR query:
Fields marked with + are also fields that can be used as labels in the SNP Tree Viewer .
-
AMR genotypes (AMR_genotypes)
Antimicrobial resistance (AMR) genes found in the isolate during analysis with AMRFinderPlus. This is a de-duplicated list, so multiple genes that share the same symbol will only be represented once. <NONE> indicates a lack of AMR genes identified by AMRFinderPlus, while an empty field means AMRFinderPlus results are not yet available. See the AMRFinderPlus analysis type, PD Ref Gene Catalog version, and AMRFinderPlus version fields for more information about the AMRFinderPlus analysis of this isolate. (Separate sections of this document provide an overview of AMRFinderPlus and additional information about genotypes.)
The genes that have been identified in an isolate's genome sequence are grouped into genotype categories, such as complete, partial, partial end of contig. The data processing pipeline section of this document provides more information about genotype categories.
Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes in the second example below), and the use of wildcards such as the asterisk and question mark (as in the first and third examples below).
Examples:- To search this field directly, enter a query such as: AMR_genotypes:searchterm
- Search for: AMR_genotypes:mcr* AND AMR_genotypes:blaKPC*
to show all of the isolates that have both a mobile colistin resistance gene and a KPC beta-lactamase. - Search for: AMR_genotypes:ant(6)-Ia AND AMR_genotypes:aph(3')-IIIa
to show all of the isolates that have both strings, ant(6)-Ia and aph(3')-IIIa, in the AMR Genotype data field. - Search for: AMR_genotypes:tet(*)
to show all of the isolates that have a genotype of "tet(*)," with the question mark serving as a wildard to retrieve gene names that have any character in the parentheses, such as tet(A), tet(M), tet(O), tet(X). etc.
-
AMR genotypes core (AMR_genotypes_core)
Core antimicrobial resistance (AMR) genes found in the isolate during analysis with AMRFinderPlus. The only differences between AMR genotypes core (AMR_genotypes_core) and AMR genotypes (AMR_genotypes) column is that "plus" genes are not shown. This is a de-duplicated list, so multiple genes that share the same symbol will only be represented once. <NONE> indicates a lack of AMR genes identified by AMRFinderPlus, while an empty field means AMRFinderPlus results are not yet available. See the AMRFinderPlus analysis type, PD Ref Gene Catalog version, and AMRFinderPlus version fields for more information about the AMRFinderPlus analysis of this isolate. (Separate sections of this document provide an overview of AMRFinderPlus and additional information about core vs. plus genotypes.)
Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes in the second example below), and the use of wildcards such as the asterisk and question mark (as in the first and third examples below).
Examples:- To search this field directly, enter a query such as: AMR_genotypes_core:searchterm
- Search for: AMR_genotypes_core:mcr* AND AMR_genotypes_core:blaKPC*
to show all of the isolates that have both a mobile colistin resistance gene and a KPC beta-lactamase.
-
AMRFinderPlus analysis type (amrfinderplus_analysis_type)
Indicates the data types that were used to analyze the isolate's genome sequences using AMRFinderPlus. Genome sequences are generally analyzed in two passes:
- NUCLEOTIDE: this in an initial analysis that is done, using translated BLAST, immediately after a pathogen isolate genome is assembled. It identifies the proteins in the genome sequence.
- COMBINED: this is a second, more sensitive analysis that runs AMRFinderPlus on both an isolate's nucleotide and protein sequences. Protein BLAST, nucleotide BLAST, and HMMER are used to analyze the proteins. The combined analysis can produce more sensitive results than the initial nucleotide analysis.
(Separate sections of this document provide details about the Pathogen Detection data processing pipeline and an overview of AMRFinderPlus. The AMRFinderPlus wiki provides details about installing and running the program, interpreting the results, and methods used.)
Data field names and values are case sensitive, as shown in the examples below, and the values of "NUCLEOTIDE" and "COMBINED" must be written in all upper case.
Examples:- To search this field directly, enter a query such as: amrfinderplus_analysis_type:searchterm
- Search for: amrfinderplus_analysis_type:COMBINED
to show all of the isolates that were analyzed by running AMRFinderPlus on both their nucleotide and protein sequences.
-
AMRFinderPlus version (amrfinderplus_version)
The version of the AMRFinderPlus software that was used to analyze a particular isolate.
New isolates are analyzed using the latest version of AMRFinderPlus software. Older isolates may have been analyzed with earlier versions of AMRFinderPlus software. There might be occasional updates to annotation on all isolates in special circumstances, such as the identification of a new genes (e.g., mobilized colistin resistance (mcr) genes).
This field will be empty if AMRFinderPlus results are not yet available.
(Separate sections of this document provide details about the Pathogen Detection data processing pipeline and an overview of AMRFinderPlus. The AMRFinderPlus wiki provides details about installing and running the program, interpreting the results, and methods used.)
Data field names and values are case sensitive (as shown in the example below, in which the data field name is in all lower case). Additional query tips are provided in a separate section of this document.
Examples:- To search this field directly, enter a query such as: amrfinderplus_version:searchterm
- Search for: amrfinderplus_version:3.6.7
to show all of the isolates that were analyzed with AMRFinderPlus version 3.6.7.
-
Assembly (asm_acc)
The accession number for the genome sequence from the Assembly database.
Data field names and values are case sensitive, as shown in the examples below.
Note that a transient state may occur where two isolates point to the same assembly when the submitter changes the taxonomic identifier for the biosample from one taxgroup to another. The assembly accession should be entered in the form of Accession.version, as in the first example below.
If you enter only the accession, no hits will be returned.
If you don't know the version number, then you can use an asterisk (*) to serve as a wildcard, as in the second example below.
In either case, the letters that are in the accession number prefix must be in upper case. A separate section of this document provides search tips about case sensitive searches.
Examples:- To search this field directly, enter a query such as: asm_acc:searchterm
- Search for: asm_acc:GCA_000008865.2
-
AST phenotypes (AST_phenotypes)
Antibiotic resistance phenotype, based on Antimicrobial Susceptibility Test (AST) results. (read more about phenotypes and look at sample records)
Data field names and values are case sensitive, as shown in the examples below. A separate section of this document provides tips about the use of quotes for phrase searches.
DISCLAIMER: Note, the format for this data field in the isolates browser is presented as a list of antibiotic compounds broken down by resistance call made by the data submitter. These are typically, done by using CLSI or EUCAST standards and those standards change over time OR the call is made by an automated instrument which may infer the cutoff. This may mean that data submitted using an earlier standard may have different resistance calls for the same antibiotic compound than data submitter using a later standard, and even for the same organism and same isolate, different tests may yield different results. Users can search this field by the antibiotic compound AND by the resistance call – the format is different than most other fields in this document.
Examples:
- To search this field directly, enter a query such as: AST_phenotypes:searchterm
- Search for: AST_phenotypes:imipenem=R
to show isolates that are resistant to imipenem - Search for: AST_phenotypes:ciprofloxacin=R OR AST_phenotypes:"nalixidic acid=R"
to show isolates that are resistant to either ciprofloxacin or nalidixic acid
- intermediate (I)
- nonsusceptible (NS)
- not defined (N, ND)
- resistant (R)
- susceptible (S, sensitive)
- susceptible-dose dependent (SSD)
-
BioProject (bioproject_acc)
BioProject accession (read more about bioprojects and look at sample records)
Data field names and values are case sensitive. The letters that are in the accession prefix must be in upper case, as shown in the example below. (A separate section of this document provides a list of accession prefixes that appear in the Pathogen Detection project.)
Examples:- To search this field directly, enter a query such as: bioproject_acc:searchterm
- Search for: bioproject_acc:PRJNA230969
to show all isolates that were sequenced as part of BioProject PRJNA230969, which describes the GenomeTrakr project by the US Food and Drug Administration (FDA) to sequence Escherichia coli (E. coli) genomes for the surveillance and rapid detection of foodborne contamination events. - Note that some bioprojects are "parent" to other bioprojects, and a search of this data field only retrieves the bioprojects that are being searched for explicitly. For exmaple, the search above will only retrieve BioProject PRJNA230969, and not its parent project (BioProject PRJNA230919). To access a parent project, or additional sub-projects that fall under the same parent, follow the "Navigate up" and "Navigate Across" links, respectively, that appear on a BioProject page.
-
BioSample (biosample_acc)
BioSample accession (read more about biosamples and look at sample records).
Data field names and values are case sensitive. The letters that are in the accession prefix must be in upper case, as shown in the example below. (A separate section of this document provides a list of accession prefixes that appear in the Pathogen Detection project.)
Examples:- To search this field directly, enter a query such as: biosample_acc:searchterm
- Search for: biosample_acc:SAMN05245394
to show the isolate from an individual BioSample, SAMN05245394, which was collected and sequenced as part of the FDA's GenomeTrakr project (BioProject PRJNA230969) for the surveillance and rapid detection of foodborne contamination events
-
Collected by (collected_by)
Name of persons or institute who collected the sample, if provided by the submitter.
Data field names and values are case sensitive, as shown in the examples below, and quotes can be used for phrase searches.
When you enter a query, the system will retrieve isolates that contain the exact query string you specified, including punctuation, capitalization, and spaces.
To browse the various values that are available in a data field, use the "Choose Columns" option at the top of the "Matched Isolates" table, select the desired column (data field) to display, then click on the column header to sort by the values in that column.
Examples:- To search this field directly, enter a query such as: collected_by:searchterm
- Search for: collected_by:FDA
-
Collection Date (collection_date)
Date sample was collected, in the format the submitter supplied.
(In contrast, the values in the Create Date field are in ISO format.)
Note: collection_date is the time the sample was collected, which may differ from the type the data was submitted to INSDC and also different than the time the data was added to the Pathogen Detection project. For real-time submissions of pathogen surveillance data, these dates will be in close proximity. For legacy data, or research projects, these dates may differ wildly and be separated by years.
You can use an asterisk (*) as a wildcard for truncation, in order to retrieve all of the isolates that were collected in a given month or year, as shown in the examples below.
To search for a range of values, enter a query such as: collection_date:[value1 TO value2] with square brackets surrounding the query string, and with the word "TO" written in upper case.
Data field names and values are case sensitive, and this data field name should be written in all lower case.
Examples:- To search this field directly, enter a query such as: collection_date:searchterm
- Search for: collection_date:2013-08-24
to show isolates in which the submitter entered that exact string as the collection date. - Search for: collection_date:2013-08
to show isolates in which the submitter entered that exact string as the collection date (that is, the isolates in which the submitter provided only the year and month, but not the day, as the collection_date). - Search for: collection_date:2013-08*
to show isolates that were collected in August 2013. The asterisk serves as a wildcard, and the system will therefore retrieve all isolates that have 2013-08 as the stem of their collection date. - Search for: collection_date:[2013-02* TO 2013-08*]
to show isolates that were collected anytime from February 2013 through August 2013.
-
Computed types (computed_types)
"In-silico" typing results. Currently the results of executing SeqSero2 on Salmonella isolates (only) are presented in these subfields:
- serotype - The serovar computed from the reads (if available) or the assembly of the isolate.
- antigen_formula - The antigenic formula computed from the reads (if available) or the assembly of the isolate.
Values for "Serotype" and "Antigen formula" in the Computed types field may not agree with the user submitted fields Serovar, TaxID, or Scientific name because those fields are reported by the submitter. The "computed_types" field, on the other hand, is a computational prediction based on the sequence calculated as part of the Pathogen Detection Pipeline.
Examples:- Search for: taxgroup_name:"Salmonella enterica" AND computed_types:serotype=Enteritidis
to show isolates whose computed serovar is Enteritidis only. - Search for: taxgroup_name:"Salmonella enterica" AND computed_types:antigen_formula=9:g,m:-
to show isolates with the antigenic formula that corresponds to serovar Enteritidis. - Search for: taxgroup_name:"Salmonella enterica" AND computed_types:serotype=Enteritidis AND NOT serovar:*nteritidis*
to show isolates whose computed serovar is Enteritidis but were submitted with a different serovar.
-
Contigs (asm_stats_n_contig)
Number of contigs in the isolate's genome assembly. If this was submitted to GenBank by the submitter it will be from their assembly and will match the assembly stats in the assembly database (https://www.ncbi.nlm.nih.gov/assembly/). If it is from an assembly made by the Pathogen Detection system, it may not yet be in GenBank, and therefore this will be the only place to see the assembly statistics.
To search for a range of values, enter a query such as: asm_stats_n_contig:[value1 TO value2]
with square brackets surrounding the query string, and with the word "TO" written in upper case. An interesting way to use a range search of this field is to retrieve isolates whose genome assemblies are comprised of only a few contigs.
Data field names and values are case sensitive, and this data field name should be written in all lower case.
Examples:- To search this field directly, enter a query such as: asm_stats_n_contig:searchterm
- Search for: asm_stats_n_contig:[1 TO 3]
to retrieve isolates with genome assemblies comprised of contigs that range in number from 1 to 3
-
Create Date (creation_date)
The date on which this isolate was first seen by the Pathogen Detection system, in the format: YYYY-MM-DD. Note, these dates are in ISO format.
(In contrast, the values in the Collection Date field are in the format that was provided by the data submitter.)
This data field does not accept an asterisk as a wild card. However, it allows you to input either a full date or a partial date as the query. For example, enter the query in the format:
YYYY-MM-DD to retrieve all isolates first seen on a specific date, orTo search for a range of values, enter a query such as: creation_date:[value1 TO value2]
YYYY-MM to retrieve all isolates first seen during a given month, or
YYYY to retrieve all isolates first seen during a given year.
with square brackets surrounding the query string, and with the word "TO" written in upper case.
Data field names and values are case sensitive, and this data field name should be written in all lower case.
Examples:- To search this field directly, enter a query such as: creation_date:searchterm
- Search for: creation_date:2013-11-19
to show isolates that were first seen by the Pathogen Detection system on that exact date. - Search for: creation_date:2013-11
to show isolates that were first seen by the Pathogen Detection system in November 2013. - Search for: creation_date:2013
to show isolates that were first seen by the Pathogen Detection system in 2013, regardless of the month or date. - Search for: creation_date:[2013-02 TO 2013-08]
to show isolates that were first seen by the Pathogen Detection system anytime from February 2013 through August 2013. - Search for: creation_date:[2013 TO 2015]
to show isolates that were first seen by the Pathogen Detection system in any month or date from 2013 through 2015.
-
Food origin (food_origin)
The geographical location where the sample originated, if provided by the submitter. This matches the /country qualifier of GenBank records. The data field typically may have two parts: Country:Region. Country is a controlled vocabulary (https://www.ncbi.nlm.nih.gov/genbank/collab/country/). Region is not controlled and can be anything (i.e., free text). For example, region could be a state abbreviation, province name, or city name.
Data field names and values are case sensitive, as shown in the examples below. If you enter both Country and Region, surround the query string in quotes. If you only specify a country and no region, then the search system will retrieve all isolates with the specified country name, regardless of region.
Note: use this field to indicate from where the sample originated, not where it was collected. Examples:- To search this field directly, enter a query such as: food_origin:searchterm
- Search for: food_origin:"USA:NY"
with quotes around the "country:region" query string, to retrieve isolates that originated in New York State. - Search for: food_origin:USA
with no space before the country name, to retrieve isolates that originated in the United States, regardless of region. (If you insert a space before the country name, the system converts the query to a search of the Text index, which is a case insensitive compilation of terms from many text-containing data fields. It will therefore retrieve isolates that contain your search term (in upper and/or lower case) in any data field.)
-
Host (host)
Host species, if provided by the submitter.
This field contains values exactly as they were entered by the data submitters. Some submitters might have entered a scientific name while others might have entered a common name; therefore, search for synonyms if you would like to retrieve more comprehensive results.
Data field names and values are case sensitive, as shown in the examples below, and a separate section of this document provides tips about using synonyms in your query.
Examples:- To search this field directly, enter a query such as: host:searchterm
- Search for: host:"Homo sapiens"
to retrieve only the isolates in which the submitter used the scientific name for the host species. - Search for: host:human
to retrieve only the isolates in which the submitter used the common name for the host species. - Search for: host:"Homo sapiens" OR host:human
to retrieve only the isolates in which the submitter used either the scientific name or the common name for the host species.
-
Host Disease (host_disease)
Host disease, if provided by the submitter.
This field contains values exactly as they were entered by the data submitters. Search for synonyms if you would like to retrieve more comprehensive results.
To browse the various values that are available in a data field, use the "Choose Columns" option at the top of the "Matched Isolates" table, select the desired column (data field) to display, then click on the column header to sort by the values in that column.
Data field names and values are case sensitive, as shown in the examples below, and separate sections of this document provides tips about using synonyms in your query, and using quotes for phrase searches.
Examples:- To search this field directly, enter a query such as: host_disease:searchterm
- Search for: host_disease:HUS
- Search for: host_disease:"hemolytic uremic syndrome"
- Search for: host_disease:"Hemolytic Uremic Syndrome"
- Search for: host_disease:HUS OR host_disease:"hemolytic uremic syndrome" OR host_disease:"Hemolytic Uremic Syndrome"
-
IFSAC_category(IFSAC_category)
IFSAC_category, if provided by the submitter. The Interagency Food Safety Analytics Collaboration (IFSAC) develops regulatory-focused schemes to help categorize isolate sourcing information.
This field contains values exactly as they were entered by the data submitters. Search for synonyms if you would like to retrieve more comprehensive results.
To browse the various values that are available in a data field, use the "Choose Columns" option at the top of the "Matched Isolates" table, select the desired column (data field) to display, then click on the column header to sort by the values in that column.
Data field names and values are case sensitive, as shown in the examples below, and separate sections of this document provides tips about using synonyms in your query, and using quotes for phrase searches.
Examples:- To search this field directly, enter a query such as: IFSAC_category:searchterm
- Search for: IFSAC_category:nuts
An alternative way to search the IFSAC_category data field is to use the "Filters" option, which includes a "IFSAC_cateogry " text box, where you can enter the category name. Here it is possible to search for null values by selecting <EMPTY>.
-
Isolate (target_acc)
Pathogen Detection accession of the isolate. The accession begins with the prefix "PDT," which stands for Pathogen Detection Target. This database is the primary resource issuing PDT accessions.
Each target is the genome assembly for a single pathogen isolate. There are several types of genome assemblies:
- isolate genomes assembled by the NCBI Pathogens data processing pipeline from sequence reads, but not published as genome sequence records in GenBank
- isolates submitted directly to GenBank as assembled genomes, and therefore have a corresponding "GCA" accession
- isolate genomes assembled by the NCBI data processing pipeline and then submitted to GenBank either by the submitter or on behalf of the submitter with their permission, or without their permission into the Third Party Annotation (TPA) database.
Data field names and values are case sensitive, and the letters that are in the accession prefix must be in upper case, as shown in the example below. (A separate section of this document provides a list of accession prefixes that appear in the Pathogen Detection project.)
The contents of this field may change for a given isolate if a new assembly or new metadata cause the pipeline to be rerun. See Data Retention and History Tracking for information on the data retention policy.
Examples:- To search this field directly, enter a query such as: target_acc:searchterm
- Search for: target_acc:PDT000133982
-
Isolate_identifiers (isolate_identifiers)
A list of alternative identifiers that the isolate may be known by.
Ids are assembled from various fields in the BioSample record, including:
- auxiliary identifiers supplied with the Biosample
- sample_name
- strain
- isolate (from BioSample)
- NARMS_isolate_number
- culture_collection
- isolate_name_alias (split by delimiter)
Data field names and values are case sensitive and embedded spaces must be contained in quotes.
Examples:- To search this field directly, enter a query such as: isolate_identifiers:searchterm
- Search for a specific identifier: CFSAN045463 isolate_identifiers:CFSAN045463
- Search for an identifier with embedded space: CVM N9107 isolate_identifiers:"CVM N9107"
- Search with a wildcard pattern: FSIS* isolate_identifiers:FSIS*
- Search a list isolate_identifiers:(PNUSAS185147 PNUSAS185148 PNUSAS185149)
-
Isolation Source (isolation_source)
Describes the physical, environmental and/or local geographical source of the biological sample from which the sample was derived, if provided by the submitter.
This field contains values exactly as they were entered by the data submitters. Data field names and values are case sensitive, as shown in the examples below. Separate sections of this document provides tips about the use of quotes for phrase searches, special characters that are part of a query term, and the use of wildcards.
Examples:- To search this field directly, enter a query such as: isolation_source:searchterm
- Search for: isolation_source:lettuce
- Search for: isolation_source:"All-Purpose Flour"
to show all isolates that have that exact string (including upper case, lower case, and hypen) in the isolation source data field. - Search for: isolation_source:*berry
to show isolates that contain terms such as strawberry, mulberry, etc. in the isolation source data field, using the asterisk as a wildcard to match zero or more sequential characters. - Note that submitters might use different terms for the same type of source (e.g., "animal-chicken-young-chicken," "chicken," "chicken breast," "Chicken Breast," "chicken carcass," "comminuted chicken," and "raw intact chicken"), so search for synonyms to broaden your retrieval, if desired.
-
Isolation type (epi_type)
Isolation type of an isolate: clinical OR environmental/other OR NULL.
Note, this field is derived from the attribute package selected by the isolate's submitter using one of the Pathogen templates in BioSample.- If attribute_package=Pathogen.cl.1.0 then isolation type is clinical.
- If attribute_package=Pathogen.env.1.0 then isolation type is environmental/other, unless host or isolation_source indicates that it was isolated from a human subject in which case isolation type is clinical.
- If neither of these packages is used then isolation type is NULL.
The isolation type (epi_type) is used to calculate the SNP distance values Min-same and Min-diff . These have non-negative values when there are other isolates in the cluster having the same or different isolation type. These values will both be n/a if the isolate has isolation type NULL. These values will also be n/a if there is no other isolate in the cluster having the same or different isolation type.
This data field's names and values are case sensitive and can be searched on values clinical OR environmental/other (enter as-is without quotes). The value NULL cannot be used as a search term. However, by using filters, you can choose between clinical OR environmental/other OR <EMPTY> and thereby find isolates whose epi_type is not set.
Examples:- To search this field directly, enter a query such as: epi_type:searchterm
- Search for clinical isolates: epi_type:clinical
- Search for environmental isolates: epi_type:environmental/other
- Search for isolates without epi_type: NOT epi_type:clinical NOT epi_type:environmental/other
-
K-mer group (kmer_group)
K-mer group accession, which is an alphanumeric representation of the Organism group. This database is the primary resource issuing PDG accession numbers. There is a one-to-one relationship of the organism group and the PDG accession, with each version representing each update of that organism group.
The K-mer accession should be entered in the form of Accession.version, as in the first example below.
If you enter only the accession, no hits will be returned.
If you don't know the version number, then you can use an asterisk (*) to serve as a wildcard, as in the second example below.
Data field names and values are case sensitive, and the letters that are in the accession prefix must be in upper case, as shown in the examples below. (A separate section of this document provides a list of accession prefixes that appear in the Pathogen Detection project.)
Examples:- To search this field directly, enter a query such as: kmer_group:searchterm
- Search for: kmer_group:PDG000000004.960
- Search for: kmer_group:PDG000000004.*
with an asterisk (*) serving as a wildcard, if you don't know the version number of the K-mer accession.
-
Lat/Lon (lat_lon)
The geographical coordinates (latitude and longitude) of the location where the sample was collected, if provided by the submitter. -
Length (asm_stats_length_bp)
Total length of the genome sequence assembly in number of base pairs (nucleotides).
If this was submitted to GenBank by the submitter it will be from their assembly and will match the assembly stats in the assembly database (https://www.ncbi.nlm.nih.gov/assembly/). If it is from an assembly made by the Pathogen Detection system, it may not yet be in GenBank, and therefore this will be the only place to see the assembly statistics.
When searching the Length data field, the value should be entered as an integer with no commas.
To search for a range of values, enter a query such as: asm_stats_length_bp:[value1 TO value2]
with square brackets surrounding the query string, and with the word "TO" written in upper case.
Data field names and values are case sensitive, and this data field name should be written in all lower case.
Examples:- To search this field directly, enter a query such as: asm_stats_length_bp:[value1 TO value2]
- Search for: asm_stats_length_bp:[4000000 TO 5000000]
to retrieve isolates with genome assemblies that are anywhere in the range of 4,000,000 to 5,000,000 nucleotides in length.
-
Level (asm_level)
Assembly level.
The NCBI Assembly database, which includes pathogen isolates as well as eukaryotic organisms, represents genomes assembled to different levels (read more about assembly levels). This field is only present for those assemblies in the assembly database. For pathogen assemblies not yet submitted to GenBank, this field will be blank, but for all intents and purposes the Pathogen Detection assemblies will only be at contig level. The Isolates Browser uses circle icons to represents the assembly levels, as follows:
- Complete Genome: Complete genome assemblies, represented in the "Level" column as a completely filled black circle icon.
- Scaffold: Assemblies that include scaffolds and contigs, represented in the "Level" column as a 1/2 filled circle icon.
- Contig: Assemblies that include only contigs, represented in the "Level" column as a 1/4 filled circle icon. Examples:
- To search this field directly, enter a query such as: asm_level:"searchterm"
- Search for: asm_level:"Complete Genome"
-
Library Layout (LibraryLayout)
Sequence Read Archive (SRA) library layout (PAIRED/SINGLE)
Data field names and values are case sensitive. The value for library layout must be entered in all upper case, as shown in the example below.
Examples:- To search this field directly, enter a query such as: LibraryLayout:searchterm
- Search for: LibraryLayout:PAIRED
-
Location (geo_loc_name)
The geographical location where the sample was collected, if provided by the submitter. This matches the /country qualifier of GenBank records. The Location data field typically may have two parts: Country:Region. Country is a controlled vocabulary (https://www.ncbi.nlm.nih.gov/genbank/collab/country/). Region is not controlled and can be anything (i.e., free text). For example, region could be a state abbreviation, province name, city name, zip code, etc.
Data field names and values are case sensitive, as shown in the examples below. If you enter both Country and Region, surround the query string in quotes. If you only specify a country and no region, then the search system will retrieve all isolates with the specified country name, regardless of region.
Note: use this field to indicate from where the sample was collected, not where it originated. Examples:- To search this field directly, enter a query such as: geo_loc_name:searchterm
- Search for: geo_loc_name:"USA:NY"
with quotes around the "country:region" query string, to retrieve isolates that were collected in New York State. - Search for: geo_loc_name:USA
with no space before the country name, to retrieve isolates that were collected in the United States, regardless of region. (If you insert a space before the country name, the system converts the query to a search of the Text index, which is a case insensitive compilation of terms from many text-containing data fields. It will therefore retrieve isolates that contain your search term (in upper and/or lower case) in any data field.)
-
Method (assembly_method)
Assembly method.
This field contains values exactly as they were entered by the data submitters.
When searching this field, the query string you enter must match exactly the string that appears in the "Method" column, including capitalization, punctuation, and spaces.
Data field names and values are case sensitive, and quotes can be used for phrase searches, as shown in the examples below.
Examples:
- To search this field directly, enter a query such as: assembly_method:"search string in quotes"
- Search for: assembly_method:"CLC NGS Cell v. 9.0"
- Search for: assembly_method:"PacBio SMRT Analysis v. 2.3.0"
- Search for: assembly_method:"SPAdes v. 3.11.1"
-
Min-same (minsame)
Minimum SNP distance from this isolate to one of the same isolation type. For example, the minimum SNP distance from one clinical isolate to another clinical isolate, or from one environmental isolate to another environmental isolate.
A value will appear in the "Min-diff" column only if an isolate has been found, by the Pathogen Detection Project data processing pipeline, to belong to a SNP cluster and another isolate in that cluster has the same isolation type (and the isolation type is not NULL). If it has, the isolate will contain a "PDS*" accession number in the "SNP cluster" column of the Isolates Browser, along with a value in the "Min-same" and/or "Min-diff" columns (depending upon the composition of the SNP cluster).
To view the SNP cluster for an isolate of interest, click on either the "PDT*" accession number in the "Isolate" column, or the "PDS*" accession number in the "SNP cluster" column. In the SNP Tree Viewer display, the branch lengths are proportional to the number of SNPs among the isolates in the cluster. Mouse over any branch to see its length.
Note that the value of Min-same is n/a where the isolate does not have a value for isolation type. It is also n/a where there are no other isolates in the cluster with this isolate's isolation type, or if the isolate is not in any SNP cluster.
To search for a range of values, enter a query such as: minsame:[value1 TO value2] with square brackets surrounding the query string, and with the word "TO" written in upper case. Data field names and values are case sensitive, and this data field name should be written in all lower case.
Examples:- To search this field directly, enter a query such as: minsame:[value1 TO value2]
- Search for: minsame:[0 TO 6]
to retrieve isolates that are no more than 6 SNPs away from other isolates of the same isolate type within the same cluster. In other words, retrieve clinical isolates that have a distance of no more than 6 SNPs from other clinical isolates in the same cluster, or retrieve environmental isolates that have a distance of no more than 6 SNPs from other environmental isolates in the same cluster.
-
Min-diff (mindiff)
Minimum SNP distance from this isolate to one of a different isolation type. For example, the minimum SNP distance from a clinical isolate to an environmental isolate, or vice versa.
A value will appear in the "Min-diff" column only if an isolate has been found, by the Pathogen Detection Project data processing pipeline, to belong to a SNP cluster and another isolate in that cluster has a different "Isolation type" that is not NULL. If it has, the isolate will contain a "PDS*" accession number in the "SNP cluster" column of the Isolates Browser, along with a value in the "Min-diff" and/or "Min-same" columns (depending upon the composition of the SNP cluster).
To view the SNP cluster for an isolate of interest, click on either the "PDT*" accession number in the "Isolate" column, or the "PDS*" accession number in the "SNP cluster" column. In the SNP Tree Viewer display, the branch lengths are proportional to the number of SNPs among the isolates in the cluster. Mouse over any branch to see its length.
Note that the value of Min-diff is n/a where the isolate does not have a value for isolation type. It is also n/a where there are no other isolates in the cluster that has a type opposite to this isolate's isolation type, or if the isolate is not in any SNP cluster.
To search for a range of values, enter a query such as: mindiff:[value1 TO value2] with square brackets surrounding the query string, and with the word "TO" written in upper case. Data field names and values are case sensitive, and this data field name should be written in all lower case. Alternatively Filters are a convenient way to search for ranges of values.
Examples:- To search this field directly, enter a query such as: mindiff:[value1 TO value2]
- Search for: mindiff:[0 to 6]
to retrieve isolates that are no more than 6 SNPs away from other isolates of the opposite isolate type within the same cluster. In other words, retrieve clinical isolates that have a distance of no more than 6 SNPs from environmental isolates in the same cluster, or vice versa.
-
N50 (asm_stats_contig_n50)
Assembly contig N50. This is a statistical measure that defines assembly quality. At least half of the bases in the assembly belong to contigs that have a length of N50 or longer.
If this was submitted to GenBank by the submitter it will be from their assembly and will match the assembly stats in the assembly database (https://www.ncbi.nlm.nih.gov/assembly/). If it is from an assembly made by the Pathogen Detection system, it may not yet be in GenBank, and therefore this will be the only place to see the assembly statistics.
When searching the N50 data field, the value should be entered as an integer with no commas.
To search for a range of values, enter a query such as: asm_stats_contig_n50:[value1 TO value2] with square brackets surrounding the query string, and with the word "TO" written in upper case. Data field names and values are case sensitive, and this data field name should be written in all lower case.
Examples:- To search this field directly, enter a query such as: asm_stats_contig_n50:[value1 TO value2]
- Search for: asm_stats_contig_n50:[1000000 TO 9999999]
to retrieve isolates with genome assemblies that are highly aggregated (in this case 50% of the assembly length is in contigs 1 Mbp or greater in size).
-
Organism Group (taxgroup_name)
Organism group related by taxonomy for purposes of calculating SNP clusters.
There is a one-to-one relationship between organism group and PDG accession. The organism group is effectively a shorthand for the organism that is predominant but does not list all organism present. These organism groups are manually constructed and may include sister species and outgroups. To see the full list of organism for each organism group utilize the scientific_name field.
Some organism groups are represented by the Genus species name, such as "Listeria monocytogenes," and others are represented as a phrase, such as "E.coli and Shigella."
Data field names and values are case sensitive, and quotes can be used for phrase searches, as shown in the example below. The system will retrieve isolates that contain the exact organism group name that you entered, including capitalization, punctuation, and spaces.
Examples:
- To search this field directly, enter a query such as: taxgroup_name:searchterm
- Search for: taxgroup_name:"Acinetobacter baumannii"
Tips:
Alternative ways to retrieve isolates that belong to a specific organism group include:
- Use the "Select an organism group" menu that appears near the top of the Isolates Browser interface, OR
- Open the complete list of Organism Groups and follow the links of interest to retrieve the isolates that belong to a group of interest.
Technical note:
- An organism group (PDG) contains one or more targets (PDTs). A PDT is a member of zero or one SNP cluster (PDS), and never more than one cluster. A SNP cluster is composed of two or more PDTs, and each PDS is completely contained within a PDG. (A separate section of this document provides a list of accession prefixes that appear in the Pathogen Detection project.)
-
Outbreak (outbreak)
The submitter designated name for an occurrence of more cases of disease than expected in a given area or among a specific group of people over a particular period of time, if provided by the submitter.
This field contains values exactly as they were entered by the data submitters.
When searching this field, the query string you enter must match exactly the string that appears in the "Outbreak" column, including capitalization, punctuation, and spaces.
Data field names and values are case sensitive, and quotes can be used for phrase searches, as shown in the example below.
Examples:- To search this field directly, enter a query such as: outbreak:"query string in quotes"
- Search for: outbreak:"1109COGX6-1 Cantaloupe"
- Search for: outbreak:"1203NYJAP-1"
- To retrieve all isolates that have a value in the outbreak data field, enter a query that uses the asterisk (wildcard) as the value.
Search for: outbreak:*
Once the search results are displayed, use the "Choose Columns" option at the top of the "Matched Isolates" table to add the "Outbreak" column to display, where you can browse the values that submitters entered in that data field.
-
PFGE Primary Enzyme Pattern (PFGE_PrimaryEnzyme_pattern)
Pulsed-field gel electrophoresis (PFGE) primary enzyme pattern, if provided by the submitter.
This field contains values exactly as they were entered by the data submitters. When searching this field, the query string you enter must match exactly the string that appears in the "PFGE Primary Enzyme Pattern" column, including capitalization and punctuation.
Data field names and values are case sensitive, as shown in the examples below.
PFGE is a DNA fingerprinting technique used to differentiate bacterial strains based on the pattern of DNA fragments that are created by digesting their complete genome with a restriction enzyme. (Read about PFGE on the CDC website and in PubMed.)
Examples:- To search this field directly, enter a query such as: PFGE_PrimaryEnzyme_pattern:searchterm
- Search for: PFGE_PrimaryEnzyme_pattern:GX6A16.0016
- Search for: PFGE_PrimaryEnzyme_pattern:JFXX01.0787
- To retrieve all isolates that have a value in the PFGE Primary Enzyme Pattern data field, enter a query that uses the asterisk (wildcard) as the value.
For example: PFGE_PrimaryEnzyme_pattern:*
Once the search results are displayed, use the "Choose Columns" option at the top of the "Matched Isolates" table to add the "PFGE Primary Enzyme Pattern" column to display, where you can browse the values that submitters entered in that data field.
-
PFGE Secondary Enzyme Pattern (PFGE_SecondaryEnzyme_pattern)
Pulsed-field gel electrophoresis (PFGE) secondary enzyme pattern, if provided by the submitter.
This field contains values exactly as they were entered by the data submitters. When searching this field, the query string you enter must match exactly the string that appears in the "PFGE Secondary Enzyme Pattern" column, including capitalization and punctuation.
Data field names and values are case sensitive, as shown in the examples below.
PFGE is a DNA fingerprinting technique used to differentiate bacterial strains based on the pattern of DNA fragments that are created by digesting their complete genome with a restriction enzyme. (Read about PFGE on the CDC website and in PubMed.)
Examples:- To search this field directly, enter a query such as: PFGE_SecondaryEnzyme_pattern:searchterm
- Search for: PFGE_SecondaryEnzyme_pattern:EXHA26.0556
- Search for: PFGE_SecondaryEnzyme_pattern:GX6A12.0022
- To retrieve all isolates that have a value in the PFGE Secondary Enzyme Pattern data field, enter a query that uses the asterisk (wildcard) as the value.
Search for: PFGE_SecondaryEnzyme_pattern:*
Once the search results are displayed, use the "Choose Columns" option at the top of the "Matched Isolates" table to add the "PFGE Secondary Enzyme Pattern" column to display, where you can browse the values that submitters entered in that data field.
-
Platform (Platform)
Sequence Read Archive (SRA) sequencing platform.
Data field names and values are case sensitive. The data field name, "Platform," should be written with a leading upper case letter, and the values are also case sensitive, as shown in the examples below.
Examples:- To search this field directly, enter a query such as: Platform:searchterm
- Search for: Platform:ILLUMINA
List of supported platforms:- ILLUMINA
- LS454
- ION_TORRENT
-
PD Ref Gene Catalog version (refgene_db_version)
The version of the Pathogen Detection Reference Gene Catalog that was used to analyze a particular isolate.
New isolates are analyzed using the latest version of the Pathogen Detection Reference Gene Catalog. Older isolates may have been analyzed with earlier versions of the Pathogen Detection Reference Gene Catalog. There might be occasional updates to annotation on all isolates in special circumstances, such as the identification of a new genes (e.g., mobilized colistin resistance (mcr) genes).
Because the "refgene_db_version" data field was added in February 2020, isolates that were analyzed prior to that time do not have a value in the corresponding "PD Ref Gene Catalog version" data column of the Isolates Browser display.
(Separate sections of this document provide details about the Pathogen Detection data processing pipeline, Pathogen Detection Reference Gene Catalog help, and an overview of AMRFinderPlus that applies the Reference Gene Catalog data in the analysis of isolate genome assemblies. The AMRFinderPlus wiki provides details about installing and running the program, interpreting the results, and methods used.)
Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes in the second example below), and the use of wildcards such as the asterisk and question mark (as in the first and third examples below).
Examples:- To search this field directly, enter a query such as: refgene_db_version:searchterm
- Search for: refgene_db_version:2020-01-06.1
to show all of the isolates that were analyzed with the Pathogen Detection Reference Gene Catalog version 2020-01-06.1. - Search for: refgene_db_version:2020-01-22.1
to show all of the isolates that were analyzed with the Pathogen Detection Reference Gene Catalog version 2020-01-22.1.
-
Run (Run)
Sequence Read Archive (SRA) accession of the sequence that was used for the genome assembly.
Data field names and values are case sensitive. The data field name, "Run," should be written with a leading upper case letter, and the "SRR" accession prefix should be written in all upper case, as shown in the examples below. (A separate section of this document provides a list of accession prefixes that appear in the Pathogen Detection project.)
Examples:- To search this field directly, enter a query such as: Run:searchterm
- Search for: Run:SRR3747659
- Search for: Run:SRR5862473 OR SRR7456389
-
Strain (strain)
Microbial strain name, if provided by the submitter.
This field contains values exactly as they were entered by the data submitters.
Data field names and values are case sensitive, as shown in the examples below.
Separate sections of this document provide tips about the use of special characters such as the hyphen, wildcards such as the asterisk, and the use of quotes for phrase searches (for strain names that contain spaces).
Examples:- To search this field directly, enter a query such as: strain:searchterm
- Search for: strain:FDA00010279
- Search for: strain:KCRI-598A
- Search for: strain::PNUSA*
-
Serovar (serovar)
Combined field of sub-species, serotype, or serovar, if provided by the submitter.
This field contains values exactly as they were entered by the data submitters.
Data field names and values are case sensitive, as shown in the examples below.
Separate sections of this document provides tips about the use of quotes for phrase searches, and special characters that appear in the sub-species, serotype, or serovar names.
Examples:- To search this field directly, enter a query such as: serovar:searchterm
- Search for: serovar:"4,[5],12:b:-"
- Search for: serovar:"Shigella sonnei"
- Search for: serovar:Enteritidis
-
SNP cluster (erd_group)
Pathogen SNP cluster accession. A SNP cluster is a group of isolates whose genome assemblies are closely related, depending on the clustering methodology used (as noted in the data processing section of this document).
The SNP cluster accession data field name is erd_group, in which "ERD" stands for Epidemiologically Related Distance.
Each SNP cluster can be viewed as a phylogenetic distance tree in the SNP Tree Viewer. (Read more in the SNP Tree Viewer help document, which includes an illustrated example of SNP Tree Viewer launch points and an illustrated example of a SNP Tree Viewer display.)
Data field names and values are case sensitive, as shown in the examples below.
The first sample search below includes an accession.version number. If you don't know the latest version number for a SNP cluster, you can use an asterisk * as a wildcard, as in the second example below. If you enter an older version number that has since been superceded by a newer version of the SNP cluster, the Isolates Browser will display a message that links to the newer version. The PDS version changes when the membership of a SNP cluster changes.
A separate section of this document provides a list of accession prefixes that appear in the Pathogen Detection project, and the data retention and history tracking section describes the use of accession.versions to track changes to the data.
Examples:
- To search this field directly, enter a query such as: erd_group:searchterm
- Search for: erd_group:PDS000003441.73
- Search for: erd_group:PDS000003441.*
with an asterisk (*) serving as a wildcard, if you don't know the version number of the SNP cluster accession. - Note: Because the SNP cluster accession is unique, it is not necessary to include the data field name in searches. It is sufficient to just enter the SNP cluster accession, if desired. For example the first search above can simply be entered as PDS000003441.73 into the Isolates Browser, and the second search can be entered as PDS000003441.*.
-
Scientific name (scientific_name)
Scientific name (in NCBI Taxonomy) of the isolate from the submitter.
Data field names and values are case sensitive, and the genus name must begin with an upper case letter. For example, enter the scientific name for: Escherichia coli. The system will retrieve isolates that have the exact string you entered. An asterisk * can be used as a wildcard, if desired.
Examples:
- To search this field directly, enter a query such as: scientific_name:searchterm
- Search for: scientific_name:"Escherichia coli O157:H7"
to retrieve the isolates containing that full, exact string as the scientific name - Search for: scientific_name:"Escherichia coli"
to retrieve the isolates containing that exact string as the scientific name, with no additional characters. - Search for: scientific_name:Escherichia*
to retrieve the isolates containing Escherichia in the scientific name, followed by any other characters.
To retrieve all isolates that belong to a specific Organism group, use the "Select an organism group" menu on the Isolates Browser home page. -
Source type (source_type)
The isolate source type. Possible values include Food, Animal, Environmental, Human, Animal feed.
Data field names and values are case sensitive, and this data field name should be written in all lower case, as shown in the example below.
Examples:
- To search this field directly, enter a query such as: source_type:searchterm
- Search for: source_type:Food
to retrieve isolates with source_type Food.
An alternative way to search the source_type data field is to use the "Filters" option, which includes a "Source type " text box, where you can enter the source_type string. Here it is possible to search for null values by selecting <EMPTY>.
-
Species TaxID (species_taxid)
The NCBI Taxonomy identifier (TaxID) at the species level for this isolate.
Data field names and values are case sensitive, and this data field name should be written in all lower case, as shown in the example below.
The TaxID number for a species can be obtained from the NCBI Taxonomy database. For example, search the database for Escherichia coli, then follow the link for that species name to open its Taxonomy Browser display, which shows a TaxID of 562.
Examples:- To search this field directly, enter a query such as: species_taxid:searchterm
- Search for: species_taxid:562
to retrieve all isolates that belong to the species Escherichia coli.
-
SRA Center (sra_center)
The name of the center that submitted the data to the Sequence Read Archive (SRA).
Data field names and values are case sensitive, as shown in the examples below.
The system will retrieve isolates that contain the exact query string you specified, including punctuation, capitalization, and spaces.
Separate sections of this document provide tips about use of quotes for phrase searches and special characters (such as a hyphen) that are part of a query term.
To browse the various values that are available in a data field, use the "Choose Columns" option at the top of the "Matched Isolates" table, select the desired column (data field) to display, then click on the column header to sort by the values in that column.
Examples:- To search this field directly, enter a query such as: sra_center:searchterm
- Search for: sra_center:EDLB-CDC
- Search for: sra_center:FDA
-
SRA Release Date (sra_release_date)
Sequence Read Archive (SRA) release date.
-
Stress genotypes (stress_genotypes)
Stress resistance genes found in the isolate during analysis with AMRFinderPlus. These can include metal, biocide, and heat resistance genes. This is a de-duplicated list, so multiple genes that share the same symbol will only be represented once. <NONE> indicates a lack of AMR genes identified by AMRFinderPlus, while an empty field means AMRFinderPlus results are not yet available. See the AMRFinderPlus analysis type, PD Ref Gene Catalog version, and AMRFinderPlus version fields for more information about the AMRFinderPlus analysis of this isolate. (Separate sections of this document provide an overview of AMRFinderPlus)
The genes that have been identified in an isolate's genome sequence are grouped into genotype categories, such as complete, partial, partial end of contig. The data processing pipeline section of this document provides more information about genotype categories.
Data field names and values are case sensitive, as shown in the examples below.
Examples:- To search this field directly, enter a query such as: stress_genotypes:searchterm
- Search for: stress_genotypes:emrE
to show all of the isolates that have the emrE gene. - Search for: stress_genotypes:emrE AND stress_genotypes:merC
to show all of the isolates that have both the emrE gene and the merC gene.
-
TaxID (taxid)
The NCBI Taxonomy identifier (TaxID) for this isolate, which can have a classification that is narrower than species.
Examples:
- To search this field directly, enter a query such as: taxid:searchterm
- Search for: taxid:83334
to retrieve isolates for Escherichia coli O157:H7.
Compare the TaxID data field that is described here with the "Species TaxID" data field that was described earlier.
The Species TaxID data field contains taxonomy IDs at the Genus species level.
The TaxID data field, in contrast, can contain classifications that are deeper than species, as shown in the examples above.
The TaxID for a species and/or for deeper nodes can be obtained from the NCBI Taxonomy database. For example, search the database for Escherichia coli, then follow the link for that species name to open its Taxonomy Browser display, which show the TaxID for the species and will list the strains that fall under it. Follow the link for any strain name of interest to open its Taxonomy Browser display and view its TaxID.
Some isolates might contain the same value in both fields, such as the E. coli isolates that are retrieved by a search for:
species_taxid:562 AND taxid:562. Those isolates have just been classified at the Genus species level, and not any deeper. -
Virulence genotypes (virulence_genotypes)
Virulence genes found in the isolate during analysis with AMRFinderPlus. This is a de-duplicated list, so multiple genes that share the same symbol will only be represented once. <NONE> indicates a lack of AMR genes identified by AMRFinderPlus, while an empty field means AMRFinderPlus results are not yet available. See the AMRFinderPlus analysis type, PD Ref Gene Catalog version, and AMRFinderPlus version fields for more information about the AMRFinderPlus analysis of this isolate. (Separate sections of this document provide an overview of AMRFinderPlus)
The genes that have been identified in an isolate's genome sequence are grouped into genotype categories, such as complete, partial, partial end of contig. The data processing pipeline section of this document provides more information about genotype categories.
Data field names and values are case sensitive, as shown in the examples below.
Examples:- To search this field directly, enter a query such as: virulence_genotypes:searchterm
- Search for: virulence_genotypes:fdeC
to show all of the isolates that have the fdeC gene. - Search for: virulence_genotypes:fdeC AND virulence_genotypes:iroE
to show all of the isolates that have both the fdeC gene and the iroE gene.
-
WGS Accession (wgs_master_acc)
The Whole Genome Shotgun (WGS) accession for the master record. The WGS master record contains no sequence data, and instead lists all of the accession numbers for the individual sequence records that compose the genome assembly for the isolate.
Tips:
The genome assembly identifier should be entered in the form of Accession.version, as in the first example below.
If you enter only the accession, no hits will be returned.
If you don't know the version number, then you can use an asterisk (*) to serve as a wildcard, as in the second example below.
Data field names and values are case sensitive, and the accession prefix must be in upper case, as shown in the examples below.
A separate section of this document provides a list of accession prefixes that appear in the Pathogen Detection project, and the data retention and history tracking section describes the use of accession.versions to track changes to the data.
Examples:
- To search this field directly, enter a query such as: wgs_master_acc:searchterm
- Search for: wgs_master_acc:JZAA00000000.1
- Search for: wgs_master_acc:JZAA00000000.*
with an asterisk (*) serving as a wildcard, if you don't know the version number of the WGS master record.
-
WGS Prefix (wgs_acc_prefix)
The stable accession prefix that is assigned to a Whole Genome Shotgun (WGS) project.
Examples:
- To search this field directly, enter a query such as: wgs_acc_prefix:searchterm
- Search for: wgs_acc_prefix:JZAA
to retrieve the isolate whose Whole Genome Shotgun (WGS) sequencing project that was assigned the prefix JZAA.
( Go back up to list of data fields or to top of document )
Search modifiers
Search modifiers can help limit the result set from a Isolates Browser search by specifying certain properties.
The available search modifiers in the Pathogens Isolates Browser include the following:
-
new
- Example: Get all the isolates added to an organism group since the last publication. Values are 0,1:
taxgroup_name:"Streptococcus pyogenes" AND new:1
-
An isolate is considered "new" when it was not included in the previous publication of the organism group. Most often a "new" isolate is represented by data newly deposited to NCBI, or added to the Pathogen Detection resource. But "new" also includes other, less common cases. These include an existing isolate was updated with new SRA data, an existing isolate was re-assembled, an isolate was dropped from Pathogen Detection then added back (there can be many reasons for this, all uncommon), or the organism group is a new one in the Pathogen Detection system.
Examples of SOLR queries
- Isolation source:
Show all isolates that have the exact phrase "All-Purpose Flour" in the isolation_source data field - Geographic location and organism group:
Show all Salmonella isolates from the USA - Genotypes: isolates that have specific genes:
Show all of the isolates that have a mobile colistin resistance gene and a KPC beta-lactamase - Phenotypes: isolates that are resistant to a given antibiotic:
Show all of the isolates that are resistant to ciprofloxacin - Genotypes and phenotypes:
Example 1: missing specific genes, resistant to antiobiotic:
Show all of the isolates that lack both a blaKPC and blaNDM carbapenemase but are resistant to imipenem
Example 2: has specific gene, susceptible to antiobiotic:
Show all of the isolates that have a blaKPC gene and are susceptible to meropenem
Example 3: has specific gene, resistant to either of two antiobiotics:
Show all of the isolates that have a qnr gene and that are resistant to either ciprofloxacin or nalidixic acid - Where is my isolate?
Retrieve your data by BioSample accession numbers or SRA Run accession numbers - Batch search with isolate identifiers that contain a hyphen:
Input a list of isolate identifiers that contain special characters (hyphens)
-
Isolation source:
Show all isolates that have the exact phrase "All-Purpose Flour" in the isolation_source data field:
isolation_source:"All-Purpose Flour"
Comments/Tips:
This query searches the "Isolation Source" data field.
Data field names and values are case sensitive and reflect the values exactly as they were entered by the data submitters. If you are uncertain about whether to use upper case, lower case, or mixed case in your query string, then you can simply enter the query in any case, but do not include a data field specifier. For example:
"all-purpose flour"
That approach searches a general Text index, which is a case insensitive compilation of terms from many text-containing data fields.
The quotes ensure that your query string will be searched as a phrase, as noted in the query tips section of the document. -
Geographic location and organism group:
Show all Salmonella isolates collected in USA:
geo_loc_name:USA AND taxgroup_name:"Salmonella enterica"
Comments/Tips:
This query searches the "Location" (geo_loc_name) and "Organism Group" (taxgroup_name) data fields.
Data field names and values are case sensitive and reflect the values exactly as they were entered by the data submitters. If you are uncertain about whether to use upper case, lower case, or mixed case in your query string, then you can simply enter the query in any case, but do not include a data field specifier. For example:
usa AND "salmonella enterica"
That approach searches a general Text index, which is a case insensitive compilation of terms from many text-containing data fields.
The quotes around the species name force the terms to be searched as a phrase. -
Genotypes: isolates that have specific genes:
Show all of the isolates that have a mobile colistin resistance gene and a KPC beta-lactamase:
AMR_genotypes:mcr* AND AMR_genotypes:blaKPC*
Comments/Tips:
This query searches the "antimicrobial resistance (AMR) genotypes" data field and uses the Boolean operator "AND" to override the default "OR."
Data field names and values are case sensitive and reflect the values exactly as they were entered by the data submitters. If you are uncertain about whether to use upper case, lower case, or mixed case in your query string, then you can simply enter the query in any case, but do not include a data field specifier. For example:
mcr* AND blakpc*
That approach searches a general Text index, which is a case insensitive compilation of terms from many text-containing data fields.
The asterisk (*) is a wild card and therefore searches for the specified word stem. -
Phenotypes: antibiotic resistance:
Show all of the isolates that are resistant to ciprofloxacin:
AST_phenotypes:ciprofloxacin=R
Comments/Tips:
The query directs the system to search the AST_phenotypes data field, where the values can be:
- I (intermediate)
- NS (nonsusceptible)
- N, ND (not defined)
- R (resistant)
- S (susceptible, sensitive)
- SSD (susceptible-dose dependent)
After you do the search, the Isolates Browser search results page will not display the "AST_phenotypes" column by default; however, you can use the "Choose Columns" option at the top of the table to add that column to the display. -
Genotypes and phenotypes:
Example 1: missing specific genes, resistant to antiobiotic:
Show all of the isolates that lack both a blaKPC and blaNDM carbapenemase but are resistant to imipenem:
AST_phenotypes:imipenem=R AND NOT AMR_genotypes:blaKPC* AND NOT AMR_genotypes:blaNDM*
- or -
AST_phenotypes:imipenem=R NOT AMR_genotypes:blaKPC* NOT AMR_genotypes:blaNDM*
Example 2: has specific gene, susceptible to antiobiotic:
Show all of the isolates that have a blaKPC gene and are susceptible to meropenem:
AST_phenotypes:meropenem=S AND AMR_genotypes:blaKPC*
Example 3: has specific gene, resistant to either of two antiobiotics:
Show all of the isolates that have a qnr gene and that are either ciprofloxacin or nalidixic acid resistant:
AMR_genotypes:qnr* AND (AST_phenotypes:ciprofloxacin=R OR AST_phenotypes:"nalixidic acid=R")
Comments/Tips:
This query searches the "antimicrobial resistance (AMR) genotypes" and "antibiotic susceptibility test (AST) Phenotypes" data fields. It also uses the Boolean operators "AND" and "NOT" to override the default "OR."
Data field names and values are case sensitive and reflect the values exactly as they were entered by the data submitters. If you are uncertain about whether to use upper case, lower case, or mixed case in your query string, then you can simply enter the query in any case, but do not include a data field specifier. For example:
imipenem=r NOT blakpc* NOT blandm*
That approach searches a general Text index, which is a case insensitive compilation of terms from many text-containing data fields.
The asterisk (*) is a wild card and therefore searches for the specified word stem. -
Where is my isolate?
Retrieve your data by BioSample accession numbers or SRA Run accession numbers:
BioSample accession number can be entered, with or without the "BioSample" (biosample_acc) data field specifier:
Sequence Read Archive (SRA) run accession number can be entered, with or without the "Run" (Run) data field specifier. If the data field specifier is used, it must begin with an upper case letter "R":
List of BioSample accession numbers, separated by white spaces, can be entered to retrieve multiple isolates, with or without the data field specifier:
List of SRA Run accession numbers, separated by white spaces, can be entered, with or without the data field specifier. If the data field specifier is used, it must begin with an upper case letter "R":
SRR3986244 SRR7294009 SRR7294010 SRR7293744 SRR7293743Mixed list of BioSample and SRA Run accession numbers, separated by white spaces, can be entered, with or without the data field specifiers, and without or with the Boolean OR:
- or -
Run:SRR3986244 SRR7294009 SRR7294010 SRR7293744 SRR7293743
-
Batch search with isolate identifiers that contain a hyphen:
Input a list of isolate identifiers that contain special characters (e.g., hyphens):
Comments/Tips:
This query retrieves the list of isolates that have the specified identifiers. The Isolates Browser has been programmed to automatically escape the special characters when they are internal to a search term, such as the dash that is part of each identifier in the list. The browser therefore interprets the special characters as part of the query string and returns isolates that contain the exact string you entered.
Output from Isolates Browser
Customize the display (choose columns, default columns, additional columns)
SNP Tree Viewer link for each isolate that belongs to a SNP cluster
Show all AMR genotypes / Hide plus AMR genotypes button
"Share" function in the Isolates Browser
Illustrated example of Isolates Browser search results
Download data from the Isolates Browser web display (metadata, assemblies)
Isolates Browser in Google Cloud BigQueryTabular list of isolates
- Upon opening the Isolates Browser, a table displays data for all available isolates, with the most recently added data at the top.
- Every row in the Isolates Browser is an assembled isolate, possibly with antimicrobial resistance (AMR), virulence, and/or stress response genotype data, and antibiotic susceptibility (AST) phenotype data, as available.
- The data for each isolate can also include strain name, geographic origin, isolation type (environmental or clinical), BioSample UID, K-mer group/organism group (PDG* accession), and more, as available. (See the Pathogens Isolates Browser data fields for a complete list.) Some of the data elements, such a accessions for corresponding BioSample and GenBank Assembly records, link to additional information in the source databases. The data in this table are either supplied by the submitter of the data into the BioProject, BioSample, SRA, and GenBank databases, and then collected from there by the Pathogen Detection system for display, or calculated by the Pathogen Detection system once the data is analyzed.
- The isolates can be sorted by clicking on column headers, faceted by using filters (e.g., Property: has antimicrobial resistance (AMR) genotypes), or searched using basic or advanced queries (see examples of SOLR queries and an illustrated example of search results).
- Tree Viewer links: If an isolate has a "PDS*" accession number in the "SNP Cluster" column, that indicates it is part of a SNP cluster, and you can click on the PSD* accession to launch the Tree Viewer and examine the relationships among your isolate of interest and other closely related isolates. read more...
Exceptions table
- The results of a search for certain isolates in an organism group may include isolates that failed quality control (QC) and so are not used for analysis. Isolates having "QC exceptions" are listed in an "Exceptions Table" along with QC details above the main grid display. Users and submitters can find out why specific isolates are not being used.
- There are three "consequences" of QC failure:
- Not published - The isolate will not appear in any published organism group (PDG).
- Not clustered - The isolate will appear in a published organism group (PDG) but will be presented as a singleton (ie no clustering attempted).
- Not submitted - The isolate will appear in a published organism group (PDG) and will be clustered, but its assembled sequence will not be submitted to GenBank.
- There are several exception "types":
- ANI species check - When aligned against a database of type strains using average nucleotide identity (ANI) on the assembled sequence, the biosample's species could not be verified.
- Readset validation failure - The SRA run was not valid and could not be used for assembly.
- Assembly validation failure - The pathogen assembly was not valid and could not be used for analysis.
- wgMLST validation failure - The GenBank assembly could not be used for clustering.
- Bad triples - The assembly failed a triangle inequality test in the legacy kmer (ie non-wgMLST) clustering step.
- The Exceptions table is published to both the Pathogen Isolates Browser and ftp. Further documentation about the ftp Exceptions file can be found at: FTP README file.
- Exception columns are defined as follows:
- exception type - The category of error
- exception - Descriptive text for this category of error
- consequence - The result of the error
- lower limit - Lower allowed limit of the value if numeric
- upper limit - Expected value, or upper limit of value if numeric
- actual value - The value of the QC check for this isolate
- BioSample - Biosample accession
- run(s) - SRA accession for the sequencing run representing this isolate.
- Isolate - Pathogen target accession for this isolate
- Assembly - GenBank assembly accession for this isolate
- organism - Organism this isolate was submitted with
- strain - Strain this isolate was submitted with
- sra center - SRA center that submitted the sequencing run
- Click the download link to download the table in comma-delimited (.csv) or tab-delimited (.tsv) format.
- Special note about assembly size validation: NCBI now validates the assembly size of most pathogenic bacterial organisms against fixed upper and lower bounds. These are set by species. The thresholds are the same for Pathogen and GenBank. The assembly size thresholds can be checked by species, see assembly size cutoffs. A table of min/max values is also available as a downloadable TSV file.
Matched clusters
- The Matched clusters window displays clusters of isolates that contain at least one isolate from the search results. Each row represents a cluster that contains one or more isolates matching the search criteria. The criteria for clustering can be found here.
- The Matched clusters columns are defined as follows:
- Organism groups- Name of the organism group. For more information about organism groups, see here.
- SNP cluster- The ID of the PDS cluster containing one or more of the isolates matching the search criteria. Clicking the hyperlink will open the cluster in the SNP Tree Viewer with the isolates matching the search criteria highlighted in red.
- Matched isolates- The number of isolates in a given cluster that match the search criteria.
- Matched clinical isolates- The number of clinical isolates in a given cluster that match the search criteria. For a definition of clinical isolates, see here.
- Matched environmental isolates- The number of environmental isolates in a given cluster that match the search criteria. For a definition of environmental isolates, see here.
- Total isolates- The total number of isolates in a cluster. Since not all isolates are assigned an Isolation Type (clinical or environmental), the total number of isolates can be larger than the sum of the matched clinical and environmental isolates.
- Minimal min-diff- Within in this cluster, the minimal number of SNPs between isolates having different isolation types, provided such isolates exist in this cluster (e.g., environmental isolates within this cluster will differ by at least X SNPs from clinical isolates or vice versa).
- Minimal min-same- Within in this cluster, the minimal number of SNPs between isolates having the same isolation type, provided such isolates exist in this cluster (e.g., environmental isolates will differ by at least X SNPs from other environmental isolates or vice versa).
- Latest update- The date that particular cluster was last updated.
Filters to refine results
- The "Filters" menu options in the Isolates Browser enable you to facet or subset the data in a variety of ways, and therefore can be used to refine your results, whether you have done a basic search or an advanced search.
- By default, each filter displays the top 100 terms (based on the number of isolates retrieved by a term) listed by count of value within that set of top 100.
- A separate section of this document provides additional information about Filters, including descriptions of the menu options, a note that filters are generated on the fly for a given data set, and tips to look for synonyms within a filter.
Sort order
- The default sort order in the Isolates Browser is by Create Date (also known as target_creation_date). That is the date on which the isolate was first seen by the Pathogen Detection system. The isolates are shown in reverse chronological order, with the newest ones appearing at the top.
- To change the sort order, click on a column header to sort by that criterion.
- Example:
- Open the Isolates Browser home page, which displays all available isolates in the default sort order.
- Enter a search for strawberr* (The asterisk is a wild card. The system therefore searches for the word stem and will retrieve isolates that contain terms such as strawberry, strawberries, etc. in any data field.)
- By default, the isolates are sorted by Create Date.
- Click on the "Organism" column header to sort alphabetically by organism name.
- Each subsequent click on the same column header inverts the sort order. (The column header acts as a toggle switch to sort in ascending or descending order by the values in that column.)
- To return to the original, default sort order, refresh the page (i.e., reload the Isolates Browser, or, if you have done a search, re-run the search).
Customize the Isolates Browser display
The options you select will persist within a given browser (e.g., Chrome, Edge, Internet Explorer, Firefox, Safari) until that browser's cookies are cleared/reset. To reset the column display and sort order to the default click the Choose columns button then click Default and OK.
SNP Tree Viewer link for each isolate that belongs to a SNP cluster
- If an isolate has a "PDS*" accession number in the "SNP Cluster" column of the Isolates Browser, this means the isolate's genome assembly has been found, via the Pathogens data processing pipeline, to be closely related to other isolate genome sequences in that SNP cluster.
- Click on either the "PDS*" (Pathogen Detection SNP Cluster) accession number or the isolate's "PDT*" (Pathogen Detection Target) accession number to open the SNP Tree Viewer, which displays an interactive phylogenetic tree of all the isolates in the SNP cluster. (A separate section of this document provides more information about the SNP Tree Viewer.)
- If the SNP Cluster column is blank for a given isolate, that means the isolate's genome assembly has not been found, by the Pathogens data processing pipeline, to be similar to any other isolate that is currently in the Pathogen Detection Project.
Show all AMR genotypes / Hide plus AMR genotypes button
- A toggle button is shown to toggle display of the AMR genotypes core or AMR genotypes column when one and only one of the two columns is shown.
"Share" function in the Isolates Browser
- A "Share" button is available in the Isolates Browser search results display. It produces a URL that captures your search strategy, which can then be copied and shared with others to execute the search. The results of the search, however, will change over time as new data become available.
Illustrated example of Isolates Browser search results

- The illustration above shows the Pathogens Isolates Browser results (as of July 24, 2018) of a search for:
escherichia AND (FDA CDC USDA) AND AST_phenotypes:*
That search retrieves isolates that contain the term "escherichia" in any data field, and contain the term FDA or CDC or USDA in any data field, and contain any value in the AST_phenotypes data field. - Click on the illustration, or enter the query above, into the Isolates Browser, to open the current, live results for that search. Once the results are displayed, use the "Choose Columns" option to customize the display, for example, by adding the "AST Phenotypes" column to the display.
- The Isolates Browser help section of this document provides additional information about searching, including basic searches, advanced searches, available data fields, and additional examples of SOLR queries.
Cross-browser selection - display isolates in MicroBIGG-E
- Selected isolates can be displayed in MicroBIGG-E, the Microbial Browser for Identification of Genetic and Genomic Elements which displays the results of AMRFinderPlus analyses.
- It is possible to view the full results in MicroBIGG-E for the isolates you have identified.
- Click the Cross-browser selection button to the right of the Expand all button (you must be logged into your myNCBI account for this functionality). By default, all of the isolates from your Isolates Browser search will be selected, as indicated by the checkbox column; however, you can deselect rows manually.
- Then click the Show in MicroBIGG-E button. A new tab will open with the MicroBIGG-E results for the selected isolates.
- For example, having identified isolates that contain a blaKPC gene and a blaTEM-1 gene, a user might want to use MicroBIGG-E to determine if these genes co-occur on the same contig. Having used the search term AMR_genotypes:blaKPC* AND AMR_genotypes:blaTEM-1, the user can click the Cross-browser selection button to the right of the Choose Columns button. By default, all of the isolates from your Isolates Browser search will be selected, as indicated by the checkbox column; however, you can deselect columns manually. Then the user can click the "Show in MicroBIGG-E" button. A new tab will open with the MicroBIGG-E results for the selected isolates.
Isolates Browser data at Google Cloud Platform in BigQuery
isolates and isolate_exceptions tables at Google BigQuery. From there the data can be analyzed and downloaded in bulk as well as linked to the microbigge table using SQL syntax.
Download data from the Isolates Browser web display
- Metadata
- Metadata can be downloaded for any isolate, whether or not it has been submitted to GenBank.
- The Isolates Browser will download the data that are currently displayed into a comma separated value (*.csv) file.
- For example, if you have chosen to customize the Isolates Browser display, only the columns you have chosen to display will be downloaded into the file.
- Bulk data in tab-delimited format per organism group can also be downloaded from the FTP site. See the ReadMe.txt on the FTP site for more information.
- To use SQL to query or to download >100,000 rows see also Isolates Browser data at Google Cloud Platform.
- Assemblies
- Assemblies can only be downloaded for isolates that have been submitted to GenBank:
- The "Assembly" column will display an accession if an isolate's assembled genome sequence has been submitted to GenBank (because assemblies that have been submitted to GenBank are also represented in the Assembly Database).
- The Assembly column will be blank if an isolate's genome sequence has not yet been submitted to GenBank. (The deposit of >500,000 isolates from the Pathogens Project into the GenBank database is an ongoing project. Many, but not all, of the isolates have been submitted to GenBank. Once the data for a given isolate have been deposited into GenBank, an accession will appear in the Assembly column, and the genomic data will be available for download at that time.)
- Assembly data are downloaded as a Generic Feature Format (GFF) file. This is a tabular 9 column file that contains the annotations generated by the Assembly Database API. The Assembly Database home page includes a link to Genomes Download FAQ, which provide more information about data downloads.
- Assemblies can only be downloaded for isolates that have been submitted to GenBank:
SNP Tree Viewer help
- What is the SNP Tree Viewer?
- RealTimeAnalysis
- How to access
- Scope of data in a tree (cutoffs for inclusion)
- Output: four panels in a SNP Tree Viewer display
- [A] Description of tree (organism group and number of isolates)
- [B] Isolates selected (navigation panel)
- [C] Table of all isolates in tree
- [D] Interactive phylogenetic distance tree
- "Neighbors" function
- "Search & Highlight in Tree"
- "Share" function
- Illustrated example of a SNP Tree Viewer display
- "Watch" function to receive automatic e-mail notifications about new data related to selected isolate(s)
- Illustrated example of an automatic notification for a watched isolate
What is the SNP Tree Viewer?
The information below provides details on real time analysis, how to access the SNP Tree Viewer, scope of data in a tree and output (four panels in a tree viewer display), which include: [A] description of tree (organism group and number of isolates), [B] isolates selected (navigation panel), [C] table of all isolates in tree, [D] interactive phylogenetic distance tree.
Real time analysis
How to access the SNP Tree Viewer
Example: The FDA's GenomeTrakr project (BioProject PRJNA230969) for the surveillance and rapid detection of foodborne contamination events include a subset of E. coli isolates that belong to the SNP cluster "PDS000003441", and that were associated with a 2016 outbreak from all-purpose flour.
In the Isolates Browser display, you can click on the "PDS*" accession number that appears in the "SNP Cluster" column for any one of those isolates to open the SNP Tree Viewer display for the SNP cluster and interactively examine the phylogenetic distance tree. (Below is an illustrated example of SNP Tree Viewer launch points.)
The resulting SNP Tree View shows a number of clinical and environmental samples that are very closely related, and therefore sheds light on the possible source of the outbreak. The SNP Tree Viewer output section of this document includes an illustrated example of a SNP Tree Viewer display that includes isolates from the E. coli outbreak. (Read more on the CDC website about that outbreak.)
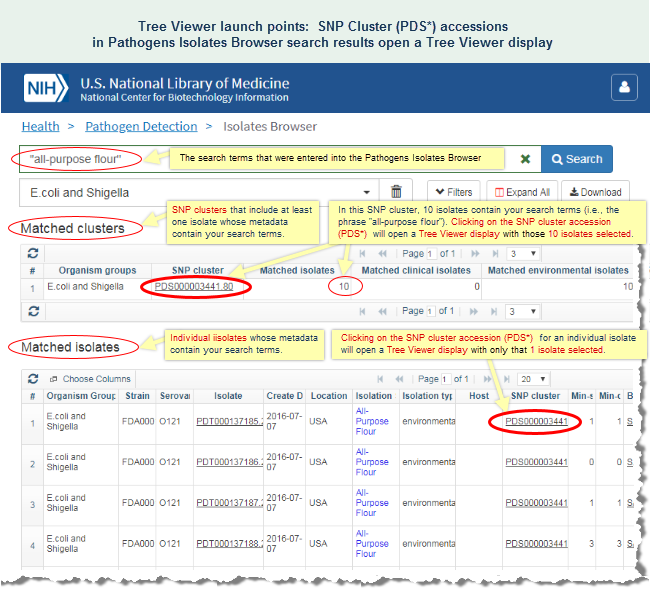
Scope of data in a tree
Individual phylogenetic trees for each SNP cluster are available on FTP as well as the NCBI Pathogen Detection Isolates Browser. (Separate sections of this file provide Isolates Browser help documentation and an overview of the data available on the FTP site.)
Output: four panels in a SNP Tree Viewer display
 Description of tree
Description of tree
- The top of a SNP Tree Viewer display provides summary information about the phylogenetic distance tree currently being displayed, such as:
- Organism group, such as E. coli and Shigella, and the corresponding PDG accession.version for the group. (The "PDG" prefix = Pathogen Detection Group.)
- Number of isolates in the tree, and the corresponding PDS accession.version for the tree. (The "PDS" prefix = Pathogen Detection SNP cluster.)
- An example is shown in part A of the illustrated example of a SNP Tree Viewer display.
- The composition of a tree can change over time as new data are added to the Pathogen Detection Project.
(A separate section of this document on data retention and history tracking provides addition information about the ways in which data and analysis results continue to evolve.)
 Isolates selected (navigation panel)
Isolates selected (navigation panel)
- The navigation panel, which has the header "Isolates Selected" in the SNP Tree Viewer interface, allows for easy tree navigation based on the selection of isolates. Clicking on ANY isolate in the navigation panel will shift the focus of the tree to where that isolate is. This is especially critical for larger trees, where the number of isolates may be several thousand, or where the number of selected isolates is large.
- The navigation panel also provides critical information on the similarity of isolates when there is more than one isolate selected, including min, max, and avg. SNP distances and the creation data ranges of the isolate(s), providing a quick and easy to use summary.
- The number of items that are listed in the "Isolates Selected" section depends upon which link you followed from the Isolates Browser output to the SNP Tree Viewer display.
- For example, the illustration of SNP Tree Viewer launch points (in the section on "how to access the SNP Tree Viewer") shows the Isolates Browser results from a search for the phrase "all-purpose flour" (as of September 4, 2018).
- In the "Matched Clusters" section of the Isolates Browser results, clicking on the Pathogen Detection SNP cluster (PDS) accession would open a SNP Tree Viewer display with "10 Isolates Selected" out of the total 136 isolates in the tree. This is because 10 of the isolates that contain your search term have been found to belong to a SNP cluster. When you view the SNP cluster in SNP Tree Viewer, those 10 isolates will be automatically selected, and will be shown in red font in the interactive phylogenetic distance tree.
- In the "Matched Isolates" section of the Isolates Browser results, clicking on an individual item (i.e., on an individual isolate's Pathogen Detection SNP cluster (PDS) or Pathogen Detection Target (PDT) accession) would open a SNP Tree Viewer display with only "1 Isolate Selected" out of the total 136 isolates in the tree.
- For example, the illustration of SNP Tree Viewer launch points (in the section on "how to access the SNP Tree Viewer") shows the Isolates Browser results from a search for the phrase "all-purpose flour" (as of September 4, 2018).
- An example of the "Isolates Selected" navigation panel is shown as part B of the illustrated example of a SNP Tree Viewer display. It features six isolates: four clinical isolates, and two environmental isolates.
- The selected isolates are also shown at the top of the table that lists all of the isolates in the SNP cluster, with their check boxes activated (as shown in part C of the illustrated example of a SNP Tree Viewer display).
- The selected isolates are displayed in red font in the phylogenetic distance tree (as shown in part D of the illustrated example of a SNP Tree Viewer display).
 Table of all isolates in tree
Table of all isolates in tree
- A table that lists all of the isolates in the SNP cluster appears above the phylogenetic distance tree. An example is shown in part C of the illustrated example of a SNP Tree Viewer display.
- The table has the same data content as the Isolates Browser, but only for the subset of isolates in the currently viewed SNP cluster. The only additional data is a checkbox column that allows selections in the table to be reflected by selections in the tree and the navigation panel. Conversely selections in the tree are reflected by selections in the table. The table can be hidden from view and customized the same as in the Isolate Browser. (A separate section of this document describes Isolates Browser output and provides information on display controls such as choose columns.)
- The "Share" button at the top of the table produces a URL that captures your customized view of the tree, which can then be shared with others to reproduce the same view. Critically, this allows the user to highlight selected isolates, collapse certain parts of the tree, and generate a view that can be shared in a document or via email with collaborators. The URL is temporary; the customized display remains available for one month. (Read more about the "share" function and data retention.)
 Interactive phylogenetic distance tree
Interactive phylogenetic distance tree
- The bottom section of a SNP Tree Viewer display shows an interactive phylogenetic distance tree, as shown in part D of the illustrated example of a SNP Tree Viewer display.
- Isolates that you have selected are shown in red font. Click on any isolate of interest in a live SNP Tree Viewer display in order to open a menu that allows you to select/deselect it.
- Display Controls above a phylogenetic distance tree in the enable you to customize the view. Mouse over a control button in a live SNP Tree Viewer display to read about its function. Some of the controls include:
- Labels button (at the top of the table that lists all of the isolates in the tree) allows you to determine which labels are displayed for the isolates in the tree view, from the set of labels that are available in the SNP Tree Viewer. The selections you make will persist within a given browser (e.g., Chrome, Edge, Internet Explorer, Firefox, Safari) until that browser's cookies are cleared/reset.
- Load Labels button allows you to add custom labels to one or more isolates in the tree view. To do this:
- On your local computer, create a tab-delimited text file (*.txt) that lists which isolates to label (by specifying their PDT* accessions), and which label(s) to add to a given isolate.
- The text file should contain one line per PDT accession and label-value pair.
- The text file can contain multiple lines with the same PDT accession. For example, if you want to add two custom labels to a given PDT, the file should contain two lines for that accession, with one label and value pair in each line.
- The contents of a sample tab-delimited text file for loading custom labels could look like:
PDT000123456 YourLabelName1 ValueA
PDT000123456 YourLabelName2 ValueB
PDT000456789 YourLabelName1 ValueC
PDT000456789 YourLabelName3 ValueD
- Save the text file on your local computer.
- Click on the "Load Labels" button and choose the file you want to load.
- A messsage will appear that says, Add N labels, where N is the number of properly formatted rows in your text file. (Each properly formatted row contains three items in a tab-delimited format: the PDT accession, a label name, and the value. If any item is missing from row, that row will not be counted, and the information it contains will not be displayed in the tree view.)
- In the case of the sample text file above, the message would say: Add 4 labels. The SNP Tree Viewer would then display ValueA and ValueB for PDT000123456, and ValueC and ValueD for PDT000456789, in addition to the other labels that were already shown for those isolates.
- Note: the Share function will not capture the custom labels you added to the display. However, you can use the "Export" option to save the customized tree in Newick, PNG, or PDF format.
- On your local computer, create a tab-delimited text file (*.txt) that lists which isolates to label (by specifying their PDT* accessions), and which label(s) to add to a given isolate.
- Expand button expands all branches (default)
- Collapse button collapses branches to show 100 nodes. Clusters with fewer nodes will not be collapsed.
- A Subtree menu appears if you click on the circle that represents a node in the tree. The Subtree menu includes options such as:
- Subtree view opens only the subtree you have selected in a new tab.
- Collapse subtree reduces the isolates in the branch into a blue cloud. Click on the collapsed node to open the menu and "Expand subtree" again, if desired.
- As an example, see part D of the illustrated example of a SNP Tree Viewer display. The lower left hand corner includes an inset showing the Subtree menu.
- The SNP Tree Viewer offers options to highlight or select groups of isolates in a single action, whether you are viewing all isolates in the tree or a only a subtree. For example:
- The "Neighbors" button (at the top of the table that lists all of the isolates in the tree) allows you to instantly select (i.e., show in red font the tree and add them to the list of "Selected isolates") all isolates that fall within a SNP distance of your originally selected isolate(s).
- "Search & Highlight in Tree" searches all labels that are currently displayed by the SNP Tree Viewer, including custom labels you might have added to the tree.
- The browser will highlight (display in bold font) isolates that contain your search term in the tree.
- The check mark icon that appears in the right hand side of the "Search & Highlight in Tree" text box allows you to select all of the highlighted isolates with a single click. Selected isolates are displayed in red font in the tree, and are added to the list of "Selected isolates" at the top of the SNP Tree Viewer display.
- If you prefer to select individual isolates, rather than the complete set of highlighted isolates, simply left click on an isolate of interest and choose "select" from the pop-up menu.
"Share" function in the SNP Tree Viewer
- A "Share" button is available in the SNP Tree Viewer display (as shown in part C of the illustrated example of a SNP Tree Viewer display). It produces a URL that captures your customized view of the tree, which can then be copied and shared with others to reproduce the same view.
- The URL is temporary, remaining valid for 60 days.
- For the first 30 days, the URL will open the customized display, showing the isolates you selected and any other customizations you made to the view.
- For the second 30 days, the URL continues to be valid, but during that time, it will only show a link to the default display for the most recent version of the SNP cluster. That is, the URL will not open the original customized view, but instead will redirect to a version of the phylogenetic distance tree that reflects the most recent for the tree.
(As noted above, under description of tree, the composition of a tree can change over time as new data are added to the Pathogen Detection Project. A separate section of this document describes the data retention and history tracking policy and examples of the ways in which data and analysis results continue to evolve.)
Illustrated example of SNP Tree Viewer display
Each tree displays all members of a SNP cluster, defined as a group of isolates whose genome assemblies are closely related, depending on the clustering methodology used (as noted in the data processing section of this document). The "Filters" option can be used, if desired, to display a subset. The interactive phylogenetic distance tree is at the bottom of a SNP Tree Viewer display, and selected isolates shown in red font in the tree.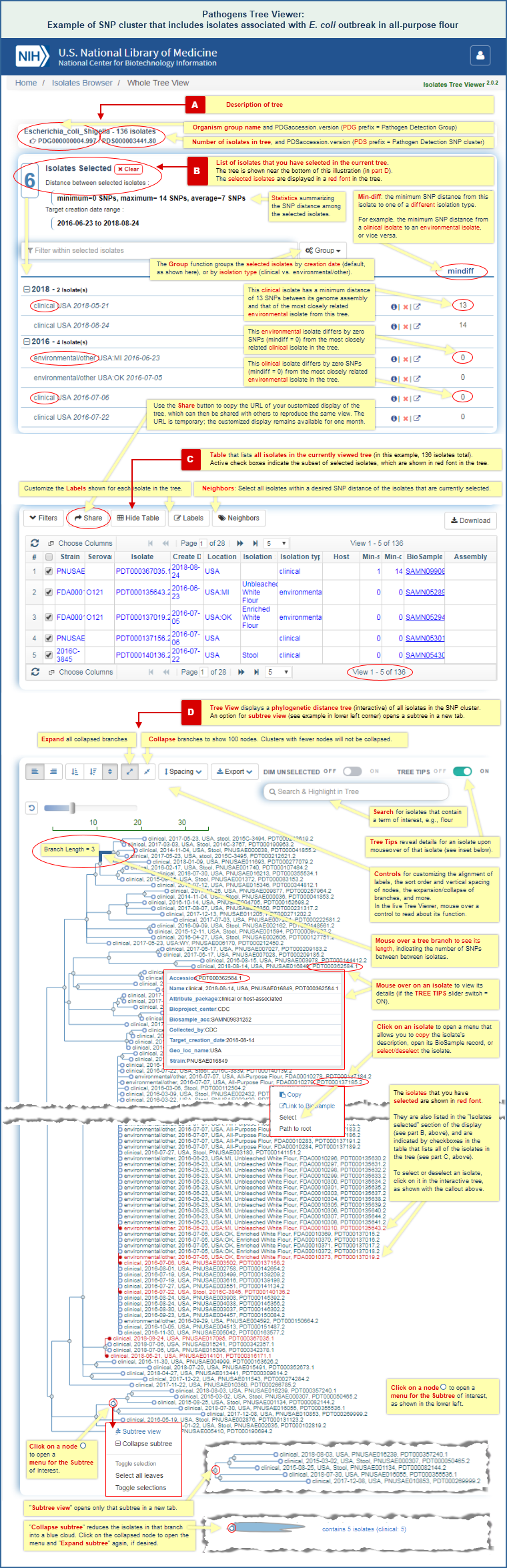
- The illustration above shows the SNP Tree Viewer display (as of September 4, 2018) for the Pathogen Detection Group (organism group) PDG000000004.997 and the SNP cluster PDS000003441.80, which includes isolates associated with an E. coli outbreak from all-purpose flour. (Read about that outbreak on the CDC website.)
- As noted above, under description of tree, the composition of a tree can change over time as new data are added to the Pathogen Detection Project.
- To open a live display of the most recent data for the SNP cluster, you can search for PDS000003441 in the Isolates Browser. That will retrieve all isolates that currently belong to that SNP cluster. Then click on the PDS000003441 accession number in the SNP Cluster column for any isolate in the search results to open the SNP Tree Viewer display for the current data. (see illustrated example of SNP Tree Viewer launch points)
- The SNP Tree Viewer help section of this document provides additional information about using the tool.
A "Share" button on the SNP Tree Viewer display can be used to copy a URL that captures your customized view of the tree, which can then be shared with others to reproduce the same view. The URL is temporary; the customized display remains available for one month (read more about the "share" function).
Automatic E-mail Notifications of New Data
- Background
- Limitations
- Requirements
- SAVE a search in the Pathogens Isolates Browser
- WATCH an isolate(s) in the SNP Tree Viewer
Background
- The NCBI Pathogen Detection Project data are updated frequently. The project includes a feature for automatic e-mail notifications of new data. It is a current awareness service to inform you about new data as it becomes available, for pathogens that are of interest to you. This feature is designed to allow users to search once, and then get automatic notifications if any pathogen isolates match their search criteria.
- Components of the automatic e-mail notifications system include:
- A "Save" button in the Isolates Browser interface,
which allows you to save a search and automatically notifies you about new isolates that match the criteria of the saved search. (Read more and view an illustrated example.) - A "Watch" button in the SNP Tree Viewer interface,
which allows you to watch one or more selected isolates in a tree, and automatically notifies you about new isolates that are similar to the isolate(s) you have chosen to watch, because they fall within the SNP distance that you have specified. (Read more and view an illustrated example.)
- A "Save" button in the Isolates Browser interface,
Limitations
- Searches are triggered for each and every organism group update that is delivered to the Pathogen Browser. An email is sent for each set of hits per organism group. That means if a search, for example for a particular antimicrobial resistance gene is not specific for a certain organism, then search results may be delivered multiple times per day. This is considered a feature and not a bug. There are currently 22 organism groups, and more are expected in the future. Not all searches can currently be done.
Requirements for automatic e-mail notifications
- My NCBI login
- Searches are tied to an email address. The only way to do this is to use your My NCBI login. If you do not yet have a My NCBI account, it is easy to set one up and there is no cost.
- You will need to be logged in to My NCBI order to save searches, which will then be run in an automated way on a daily basis. The system will send e-mail notifications when new data arrive for a saved search.
- You do not need to be logged in to receive the e-mail notifications. The notifications will be sent to the My NCBI email address you used when creating the account.
- More information about My NCBI is available in the My NCBI help document, video overview (YouTube).
- The main function of MyNCBI for the Pathogens Isolates Browser is to associate your e-mail address with the searches that you save, so you can received e-mail notifications about new data.
- The My NCBI help document and video overview, above, provide general information about My NCBI and are included here as a general reference.
- Some of the features described in help document and video overview apply to NCBI databases that are within the Entrez search system, but might not apply to Pathogens, which is outside of that system because it uses a different search engine (SOLR).
- For example, the Pathogens saved searches will not appear directly on your My NCBI account page, but are instead accessible through the "Saved Searches" link in the Pathogens Isolates Browser or the "Watched Isolates" link in the SNP Tree Viewer.
- Perform search in Pathogens Isolates Browser
- In order to received automated search results by e-mail, a search first needs to be performed in the Pathogens Isolates Browser.
- We recommend starting with narrowly defined searches, otherwise you will be inundated with meaningless and noisy search results by email, which would be better performed in the browser when needed.
(A separate section of this document provides Isolates Browser help, which includes detailed information about search syntax, including allowable search terms, query tips, advanced search techniques that use the SOLR query language, a complete list of data fields, and examples of SOLR queries.) - Then you can either SAVE your search and/or WATCH an isolate(s) of interest, as described below, in order to receive automatic e-mail notifications of any new data that become available.
SAVE a search in the Pathogens Isolates Browser
- Your search will then be run in an automated way on a daily basis .
- You will receive automatic e-mail notifications only if/when new isolates become available that match your search criteria.
- Use the "Saved Searches" link on the Pathogens Isolates Browser interface to view the list of your saved searches, and to edit or delete the searches.
- The illustrated example below shows the "Save" button, the "Saved Searches" link, and a sample automatic e-mail for a saved Search.
Illustrated example of automatic e-mail notification for a Saved Search
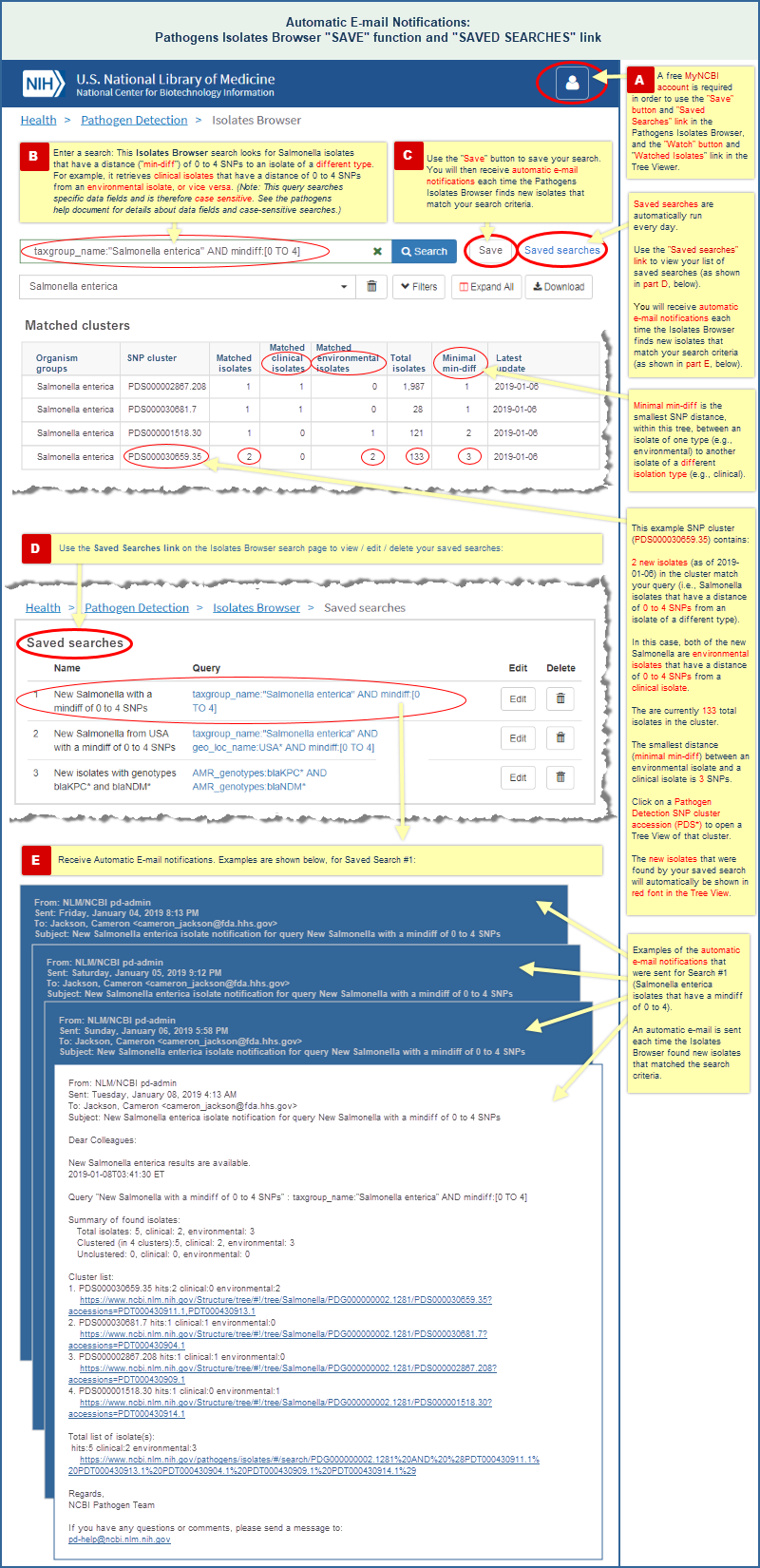
WATCH an isolate in the SNP Tree Viewer
- If you select multiple isolates in the SNP Tree View and then press the "Watch" button, then all of the selected isolates will be added to your list of watched isolates.
- The system will prompt you to enter a name for the watched isolate(s), and to specify the maximum SNP distance for receiving reports of new data.
- Each isolate will be watched on a daily basis in an automated way.
- You will receive automatic e-mail notifications only if/when new isolates that fall within a specified SNP distance of the isolate(s) that you select in that tree view.
- Use the "Watched Isolates" link on the SNP Tree Viewer interface to view your list of watched isolates, and to rename a watch, edit the SNP cutoff, or delete it from your list.
- The illustrated example below shows the "Watch" button, the "Watched Isolates" link, and a sample automatic e-mail for a watched isolate.
Illustrated example of automatic e-mail notification for a Watched Isolate
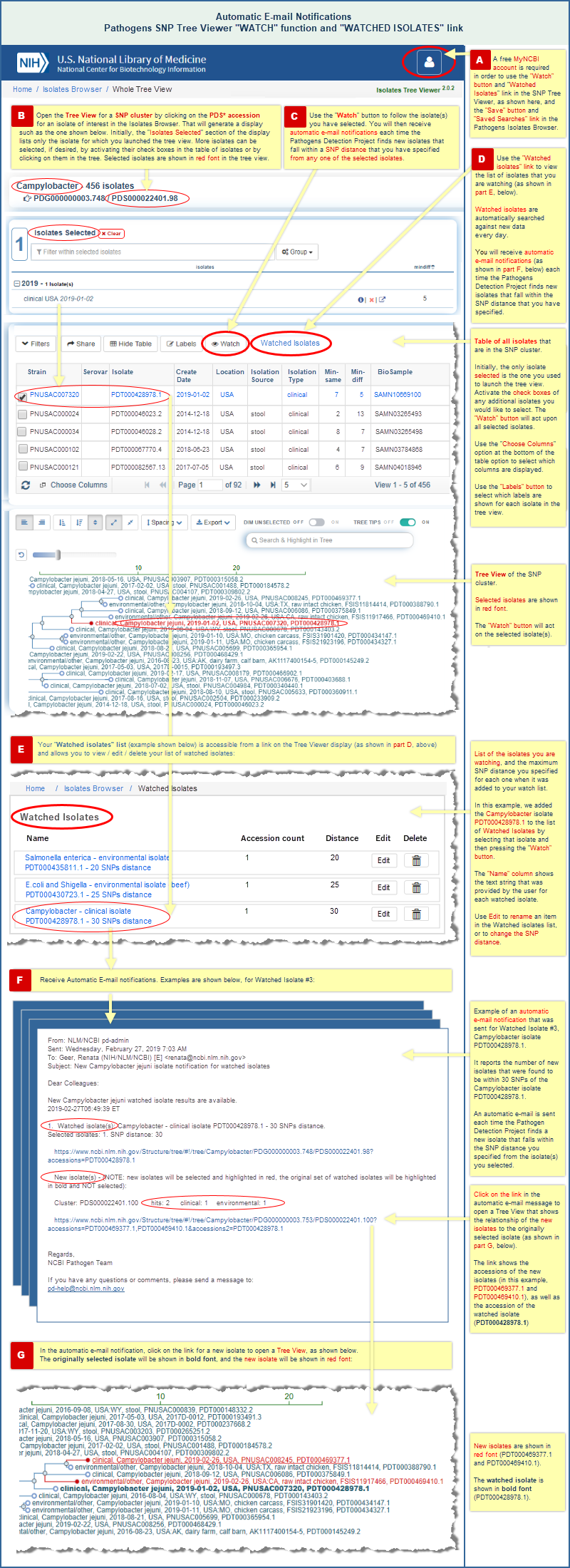
Antimicrobial Resistance (AMR) Resources
- Overview (schematic illustration of AMR resources)
- AMR Landing page
- AMR Resources page
- Pathogen Detection Reference Gene Catalog
- AMRFinderPlus
- MicroBIGG-E (Microbial Browser for Identification of Genetic and Genomic Elements)
- Submit sequence and phenotype data related to AMR
- FTP/Raw Data Download
AMR Overview
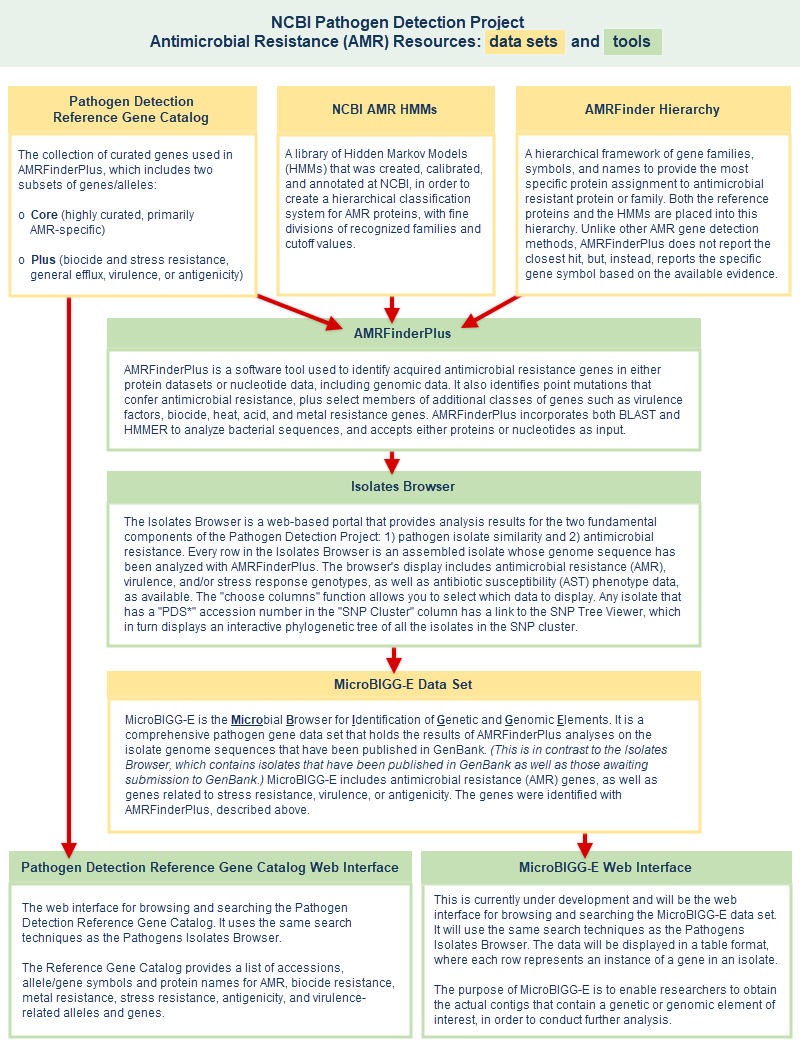
- Pathogen Detection Reference Gene Catalog: read more, browse/search, download data, Change log
- NCBI AMR Hidden Markov Models (HMMs): read more, browse/search, download data
- AMRFinder Hierarchy: read more, read more, view/download file
- AMRFinderPlus: read more, install
- Isolates Browser: read more about the Isolates Browser, about genotypes (which are displayed in the AMR_genotypes, stress_genotypes, and virulence_genotypes data fields); and about phenotypes (which are displayed in the AST_phenotypes data field and are based upon Antimicrobial Susceptibility Test (AST) results)
- MicroBIGG-E (Microbial Browser for Identification of Genetic and Genomic Elements): read more, browse/search
AMR Landing page
AMR Resources page
Pathogen Detection Reference Gene Catalog help
- What is the Pathogen Detection Reference Gene Catalog?
- Where to access the Pathogen Detection Reference Gene Catalog
- Search tips
- Data Fields
- allele
- gene_family
- product_name
- scope
- type
- subtype
- class
- subclass
- refseq_protein_accession
- refseq_nucleotide_accession
- genbank_protein_accession
- genbank_nucleotide_accession
- organism fields:
- curated_refseq_start
- genbank_start
- genbank_stop
- genbank_strand
- refseq_start
- refseq_stop
- refseq_strand
- pubmed_reference
- synonyms
- links
- Output
- Use cases/sample searches of the Reference Gene Catalog
What is the Pathogen Detection Reference Gene Catalog?
Every row in the Pathogen Detection Reference Gene Catalog display is a reference gene or a point mutation.
Scope: the Reference Gene Catalog includes two data subsets:
- "Core": this subset includes highly curated, AMR-specific genes and proteins from the Bacterial Antimicrobial Resistance Reference Gene Database (BioProject PRJNA313047), plus point mutations. The sources of input for this curated database include: 1) allele assignments, 2) exchanges with other external curated resources, 3) reports of novel antimicrobial resistance proteins in the literature.
- "Plus": this subset includes genes related to biocide and stress resistance, general efflux, virulence or antigenicity, or other AMR genes whose presence/absence are unlikely to affect phenotype and/or whose phenotype is highly uncertain.
Proteins in the reference gene catalog may be recategorized from core to plus or vice-versa based on curation and the literature. Changes are enumerated in the 'changes.txt' file for each AMRFinderPlus database release.
- The definition of redundant (or 'non-unique') will differ, depending on the type of data element (allele, gene, or point mutation). For example:
- An ALLELE should only ever show up once in the table. An allele is a unique protein sequence that corresponds to a unique gene symbol, and so, by definition, should occur only once.
- An allele name for a POINT MUTATION can occur in multiple rows of the Reference Gene Catalog, if the point mutation is found in different organisms, and if the proteins in those organisms are not identical.
-
For example, the allele name gyrA_D82G occurs in both E. coli and Salmonella. Each of those organisms has its own reference sequence protein (WP_* accession), because the protein sequences are not identical. The E. coli gyrA protein sequence is WP_001281243.1, and the Salmonella gyrA protein is WP_001281271.1.
- If, on the other hand, two or more organisms have an identical protein sequence for a given gene, and the same allele has been found in all of those organisms, there will be a single row in the Reference Gene Catalog, showing the allele name and the Reference Sequence WP_* accession.
-
For example, the allele name gyrA_D82G occurs in both E. coli and Salmonella. Each of those organisms has its own reference sequence protein (WP_* accession), because the protein sequences are not identical. The E. coli gyrA protein sequence is WP_001281243.1, and the Salmonella gyrA protein is WP_001281271.1.
- A given GENE SYMBOL can have multiple rows in the table, as multiple proteins can be assigned the same gene symbol, but each WP_* accession will be unique.
Details about WP_* accessions are provided on the web pages that describe the RefSeq non-redundant proteins, the Prokaryotic RefSeq Genome Re-annotation Project, and the New RefSeq protein product and data model.
- The Pathogen Detection Reference Gene Catalog and Pathogens Isolates Browser are related resources and are integrated with each other.
- The main similiarities between the resources are their shared search engine and similar search techniques:
- Both use the SOLR query language and allow searches by a wide variety of text terms.
- The search tips provided in the Isolates Browser help documentation therefore also apply to the Reference Gene Catalog, such as basic search techniques, advanced search techniques, case sensitive versus case insensitive searches, and the availability of "filters" to refine search results.
- The main differences between the resources are the scope of data being searched, the set of data fields (and filters, which are based on data fields) that are available for searching, and the columns that are shown in the display of search results:
- The Pathogens Isolates Browser searches all isolate genomes in the Pathogen Detection project, as well as corresponding metadata that are described under types of data.
- Every row in the Isolates Browser is an assembled isolate, possibly with antimicrobial resistance (AMR), virulence, and/or stress response genotype data, and antibiotic susceptibility (AST) phenotype data, as available.
- The Isolates Browser help documentation describes the available data fields and output.
- The Pathogen Detection Reference Gene Catalog is a non-redundant database of bacterial genes related to antimicrobial resistance, biocide and stress resistance, general efflux, virulence, or antigenicity.
- Every row in the Pathogen Detection Reference Gene Catalog display is a reference gene or a point mutation.
- The Reference Gene Catalog help describes the scope of data, available data fields, and output.
- The Pathogens Isolates Browser searches all isolate genomes in the Pathogen Detection project, as well as corresponding metadata that are described under types of data.
- They are integrated with each other through links in the data displays. For example:
- The gene family column in the Pathogen Detection Reference Gene Catalog links to the Isolates Browser. Clicking on a gene symbol in that column will open the Isolates Browser and will display the isolates that have been found, by the data processing pipeline, to contain the gene of interest.
- The Pathogen Detection Reference Gene Catalog, Reference Gene Hierarchy, and Reference HMM Catalog are interrelated and integrated databases that are all used in concert to identify gene sequences by AMRFinderPlus.
Curation of Reference genes and HMMs is organized in the framework of the Reference Gene Hierarchy, so looking at this resource will show how Pathogen Detection curators relate genes to one-another. Note that genes are placed in the Reference Gene Hierarchy, but point mutations are not. - As with the isolates browser all use a shared search engine and similar search techniques:
- All use the SOLR query language and allow searches by a wide variety of text terms.
- The search tips provided in the Isolates Browser help documentation therefore also apply to the reference browsers, such as basic search techniques, advanced search techniques, case sensitive versus case insensitive searches, and the availability of "filters" to refine search results.
- To link out to the Reference Gene Hierarchy from the Reference Gene Catalog use the Choose columns button to add the Hierarchy node ID column to the table then click on the Hierarchy node ID you wish to see in the Reference Gene Hierarchy
Where to access the Pathogen Detection Reference Gene Catalog
Browse/Search the Reference Gene Catalog:
https://www.ncbi.nlm.nih.gov/pathogens/refgene.
Download Reference Gene Catalog data:
Data from the Reference Gene Catalog can be downloaded in multiple formats. From the web interface you can get sequence and table data you see by clicking on the Download button at the top of the table (See the Output section for more info).
To get the data in table format click Download then select the File type: Table, select either tab-delimited (.tsv) or comma-delimited (.csv) and select a filename to download. Only the rows and columns that are visible in the table view on the web interface will be included in the downloaded file.
To get sequence data from the web interface click the Download button then select the File type: Dataset. Choose Reference nucleotide, Reference nucleotide with flanks, and/or the Reference protein sequence to download in FASTA format. Note that reference sequences for point mutations will be the "wildtype" references not including the mutations, and that RNA genes or promoter region references will not have protein sequences. Flanking nucleotide sequences may be limited to 100-bp or less depending on the source sequences in GenBank or RefSeq. The .zip file downloaded will be in the "Datsets" format including the metadata for sequences included in JSON format. See the NCBI Datasets documentation for more information on metadata file formats.
A tab-delimited table of the metadata in the Reference Gene Catalog is available in the AMRFinderPlus database release. See the AMRFinderPlus database documentation for the location and the ReferenceGeneCatalog.txt file format documentation for more information.
Search tips for the Pathogen Detection Reference Gene Catalog
- The Pathogen Detection Reference Gene Catalog can be searched by the terms that appear in any of the data fields described below. A search example is provided after each data field description, when possible.
- The query tips described in the Isolates Browser help > basic search section also apply to the Reference Gene Catalog, such as searches for multiple terms, special characters, phrase searches, case sensitive vs. case insensitive searches, etc.
- The query tips described in the Isolates Browser help > advanced search section also apply to the Reference Gene Catalog, because both resources use the SOLR query language.
- The main difference is the data fields that are available to be searched, because each resource has its own set of data fields. (See a list of the data fields in the Pathogen Detection Reference Gene Catalog (below) or the data fields in the Isolates Browser.)
- The query tips described in the Isolates Browser help also apply to the Reference Gene Catalog, such as searches for multiple terms, special characters, phrase searches, case sensitive vs. case insensitive searches, etc.
- The "Filters" menu options in the Pathogen Detection Reference Gene Catalog enable you to facet or subset the data in a variety of ways, and therefore can be used to refine your results, whether you have done a basic search or an advanced search.
- By default, each filter displays the top 100 terms (based on the number of items retrieved by a term) listed by count of value within that set of top 100. Note that:
- A Boolean "OR" is applied if multiple items are checked in the same filter field. This way you can choose multiple values in the same filter. For example:
- Open the "Filters" tab of the Pathogen Detection Reference Gene Catalog, then check the boxes for "Stress" and for "Virulence" in the "Type" filter. The system will retrieve genes that are associated with either stress resistance or with virulence.
- A Boolean "AND" is applied if you select items in several different filter fields (Type, Class, etc). For example:
- Open the "Filters" tab of the Pathogen Detection Reference Gene Catalog, then check the boxes for "Point" in the "Subtype" filter and "Quinolone" in the "Class" filter. The system will retrieve alleles that meet both of your specified criteria (in this case, point mutations that confer resistance to quinolones).
- A Boolean "OR" is applied if multiple items are checked in the same filter field. This way you can choose multiple values in the same filter. For example:
- As explained in the Isolates Browser help, Filters are generated on the fly. As a result, the terms that are listed under each filter will depend on the data set you are currently displaying in the browser. That is also true for the filters in the Pathogen Detection Reference Gene Catalog.
Data Fields in the Pathogen Detection Reference Gene Catalog
Each data field reflects an available column in the Pathogen Detection Reference Gene Catalog web interface. The output section of this document provides tips on how to customize the display, using the "choose columns" function.
Please note: in the list of available data fields below:
- The term shown in the regular font is the display name (column header) shown by the Pathogen Detection Reference Gene Catalog web interface. The term shown in (italics) is the name of the corresponding data field, if you want to search that field directly.
- For example, one data field is listed as: gene family (gene_family). The phrase "gene family" (with a space between the words) appears in the Reference Gene Catalog column header, and "gene_family" (with an underscore bar instead of a space) is the string you should use if you want to search that data field directly.
- Brief italicized search examples are also provided for each data field, when possible, showing how to query the data field directly. The values represent text strings exactly as they appear in the data fields, including upper case and lower case letters, special characters such as hyphens, etc. The data field names and values are case sensitive.
Note that each field is written in this format: display name (data_field_name)
The "display name" is the column header that appears in the Reference Gene Catalog web interface, and the "data_field_name" is the case-sensitive string you should enter if you want to search the data field directly using a SOLR query:
-
Allele (allele)
Gene or allele. If the data element is an allele (e.g., 23S_C2627A), its name reflects both the name of the gene family in which a point mutation was found, and the location coordinate of the mutation, and the wild type and mutated nucleotides/amino acids
Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: allele:searchterm
- Search for: allele:gyrA_D82G
to show all alleles with that name.
(A separate section of this document describes the non-redundant nature of the Reference Gene Catalog, and how the definition of redundant (or 'non-unique') will differ, depending on the type of data element (allele, gene, or point mutation).) - Search for: allele:blaB-1
to show the reference gene for the blaB-1 allele: subclass B1 metallo-beta-lactamase BlaB-1. - Search for: allele:blaB-*
to show the reference genes for all blaB alleles.
-
Gene family (gene_family)
Gene symbol, or, if a point mutation, the reference gene symbol.
Data field names and values are case sensitive. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: gene_family:searchterm
- Search for: gene_family:bla2
to show members of the bla2 gene family: BcII family subclass B1 metallo-beta-lactamases. Each hit will correspond to a unique protein sequence, and corresponding unique nucleotide sequence. That is, each hit will have a unique WP_* accession (refseq_protein_accession), and/or a corresponding unique NG_* accession (refseq_nucleotide_accession). (A separate section of this document describes the non-redundant nature of the Reference Gene Catalog, and how the definition of redundant (or 'non-unique') will differ, depending on the type of data element (allele, gene, or point mutation).)
-
Product name (product_name)
Name of gene product or genomic region.
Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of quotes to search for a phrase.
Examples:- To search this field directly, enter a query such as: product_name:searchterm
- Search for: product_name:"BcII family subclass B1 metallo-beta-lactamase"
to show all entries in the Reference Gene Catalog that have the exact product name that you specified, including upper and lower case letters as well as special characters (in this case, hyphens). As of July 5, 2019, the search retrieves 6 hits.
Note: If the search is entered without quotes surrounding the product name, such as:
Search for: product_name:BcII family subclass B1 metallo-beta-lactamase then each space is interpreted by the search system as a Boolean OR. As of July 5, 2019, the search retrieves 1,466 hits.
(read more about SOLR operators)
-
Scope (scope)
This field specifies the data subset to which an allele or gene belongs, and the value can either be core (curated for relevance to resistance, usually AMR-specific genes and point mutations) or plus (genes related to biocide and stress resistance, general efflux, virulence, or antigenicity , or where the presence of this gene may not be informative as to resistance phenotype or the relationship is not clear).
Data field names and values are case sensitive. In this case, both the data field name and the value are written in all lower case, as shown in the example below.
Examples:- To search this field directly, enter a query such as: scope:searchterm
- Search for: scope:plus
to show the genes in the "plus" subset of the Pathogen Detection Reference Gene Catalog. That subset includes genes related to biocide and stress resistance, general efflux, virulence, or antigenicity.
-
Type (type)
Classification for the type of gene found, such as AMR, STRESS, or VIRULENCE. A more detailed description of the type and subtype fields is available on the AMRFinderPlus wiki
(In general, type and subtype refer to the category of gene or genetic element, while class and subclass refer to the a phenotype associated with the genetic element.)
Data field names and values are case sensitive, and the values for this data field are written in upper case, as shown in the example below.
Examples:- To search this field directly, enter a query such as: type:searchterm
- Search for: type:STRESS
to show genes that confer stress resistance.
As an alternative method for retrieving those genes, you can open the "Filters" function of the Reference Gene Catalog and check the box for the desired Type. By doing so, the Filters function will refresh itself to show the subtype values that are available for the type you have selected, enabling you to further narrow your search results, if desired. For example, the subtype values under STRESS currently include BIOCIDE, HEAT, and METAL. (As noted below, filters are generated on the fly and reflect the attributes of the data that you are currently viewing.)
-
Subtype (subtype)
Classification for the subtype of gene found. A more detailed description of the type and subtype fields is available on the AMRFinderPlus wiki
(In general, type and subtype refer to the category of gene or genetic element, while class and subclass refer to the substrate.)
Data field names and values are case sensitive, and the values for this data field are written in upper case, as shown in the example below.
Examples:- To search this field directly, enter a query such as: subtype:searchterm
- Search for: subtype:HEAT
to show genes that confer heat resistance.
As an alternative method for retrieving those genes, you can open the "Filters" function of the Reference Gene Catalog and check the box for the desired Subtype. By doing so, the Filters function will refresh itself to show the corresponding type under which the selected subtype falls. For example, the subtype value of HEAT falls under the type STRESS. (As noted below, filters are generated on the fly and reflect the attributes of the data that you are currently viewing.)
-
Class (class)
"Class" provides a broad definition of the phenotype affected by the gene or allele, and includes resistance phenotypes such as antimicrobial and stress resistance, virulence, and antigenicity. For some virulence genes this field contains typing information. More information about class and subclass fields can be found on the AMRFinderPlus wiki
(In general, type and subtype refer to the category of gene or genetic element, while class and subclass refer to the substrate.)
Data field names and values are case sensitive, and the values for this data field are written in upper case, as shown in the example below.
Additional sections of this document provide tips search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: class:searchterm
- Search for: class:BETA-LACTAM
to show all genes classified as BETA-LACTAM.
As an alternative method for retrieving those genes, you can open the "Filters" function of the Reference Gene Catalog and check the box for the desired Class. By doing so, the Filters function will refresh itself to show the subclass values that are available for the type you have selected, enabling you to further narrow your search results, if desired. For example, the subclass values under BETA-LACTAM currently include BETA-LACTAM, CARBAPENEM, CEPHALOSPORIN, CEPHALOTHIN, and METHICILLIN. (As noted below, filters are generated on the fly and reflect the attributes of the data that you are currently viewing.)
-
Subclass (subclass)
Where it is known, "Subclass" provides a more specific definition of the particular antibiotics or classes that are affected by the gene or point mutation (e.g., that are resisted by the gene/allele). While most subclass designations are self-explanatory, a few others have particular meanings. Specifically, "CEPHALOSPORIN" is equivalent to the Lahey 2be definition; "CARBAPENEM" means the protein has carbapenemase activity, but it might or might not confer resistance to other beta-lactams; "QUARTERNARY AMMONIUM" are quarternary ammonium compounds. In addition, stx subtypes (e.g., STX2E) and intimin subtypes (e.g., ALPHA) are defined for Shiga toxin proteins (class of STX1 or STX2) and intimins (class of INTIMIN) respectively. Where the phenotypic information is incomplete, contradictory, or unclear, the "Class" value is used for the "Subclass" value.
More information about the class and subclass fields can be found on the AMRFinderPlus wiki
(In general, type and subtype refer to the category of gene or genetic element, while class and subclass refer to the substrate.)
Data field names and values are case sensitive, and the values for this data field are written in upper case, as shown in the example below.
Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: subclass:searchterm
- Search for: subclass:CEPHALOSPORIN
to show genes that confer resistance to cephalosporin antibiotics.
As an alternative method for retrieving those genes, you can open the "Filters" function of the Reference Gene Catalog and check the box for the desired subclass. The Filters function will then refresh itself to show the corresponding class under which the selected subclass falls. For example, the subclass value of CEPHALOSPORIN falls under the class BETA-LACTAM. (As noted below, filters are generated on the fly and reflect the attributes of the data that you are currently viewing.)
-
RefSeq protein accession (refseq_protein_accession)
Accession of the RefSeq protein sequence record in which the gene or allele is found. It generally has a WP_* prefix. (Read more about RefSeq, the distinct format of RefSeq accessions, and the various accession prefixes that appear in the Pathogen Detection project.)
Enter the sequence record identifier in the accession.version format, as shown in the first example below.
If you don't know the version of the sequence record, enter just the accession, as shown in the second example below.
Examples:- To search this field directly, enter a query such as: refseq_protein_accession:searchterm
- Search for: refseq_protein_accession:WP_001281243.1
to show the Reference Gene Catalog entries associated with this RefSeq protein sequence record. If multiple alleles have been found to exist in this protein, there will be a separate entry for each allele. (A separate section of this document describes the non-redundant nature of the Reference Gene Catalog, and how the definition of redundant (or 'non-unique') will differ, depending on the type of data element (allele, gene, or point mutation.) - Search for: refseq_protein_accession:WP_001281243
to show the Reference Gene Catalog entries associated with this RefSeq protein accession, regardless of its version number. (The Reference Gene Catalog contains the latest version of a sequence record; if you don't know what version number is the latest, enter only the accession as your query, without any dot or version number.)
-
RefSeq nucleotide accession (refseq_nucleotide_accession)
Accession of the RefSeq nucleotide sequence record in which the gene or allele is found. It generally has an NG_* prefix. (Read more about RefSeq, the distinct format of RefSeq accessions, and the various accession prefixes that appear in the Pathogen Detection project.)
Enter the sequence record identifier in the accession.version format, as shown in the first example below.
If you don't know the version of the sequence record, enter just the accession, as shown in the second example below.
Examples:- To search this field directly, enter a query such as: refseq_nucleotide_accession:searchterm
- Search for: refseq_nucleotide_accession:NG_047553.1
to show the Reference Gene Catalog entry associated with this RefSeq nucleotide sequence record. - Search for: refseq_nucleotide_accession:NG_047553
to show the Reference Gene Catalog entries associated with this RefSeq nucleotide accession, regardless of its version number. (The Reference Gene Catalog contains the latest version of a sequence record; if you don't know what version number is the latest, enter only the accession as your query, without any dot or version number.)
-
GenBank protein accession (genbank_protein_accession)
Accession of the GenBank protein sequence record in which the gene or allele is found. (Read more about the format of GenBank accessions, and about the various accession prefixes that appear in the Pathogen Detection project.)
Enter the sequence record identifier in the accession.version format, as shown in the example below.
If you don't know the version of the sequence record, enter just the accession, as shown in the second example below.
Examples:- To search this field directly, enter a query such as: genbank_protein_accession:searchterm
- Search for: genbank_protein_accession:AAB00464.1
to show the Reference Gene Catalog entries associated with this GenBank protein. - Search for: genbank_protein_accession:AAB00464
to show the Reference Gene Catalog entries associated with this GenBank protein accession, regardless of its version number. (The Reference Gene Catalog contains the latest version of a sequence record; if you don't know what version number is the latest, enter only the accession as your query, without any dot or version number.)
-
GenBank nucleotide accession (genbank_nucleotide_accession)
Accession of the GenBank nucleotide sequence record in which the gene or allele is found. (Read more about the format of GenBank accessions, and about the various accession prefixes that appear in the Pathogen Detection project.)
Enter the sequence record identifier in the accession.version format, as shown in the example below.
If you don't know the version of the sequence record, enter just the accession, as shown in the second example below.
Examples:- To search this field directly, enter a query such as: genbank_nucleotide_accession:searchterm
- Search for: genbank_nucleotide_accession:L26954.1
to show the Reference Gene Catalog entries associated with this GenBank nucleotide sequence. - Search for: genbank_nucleotide_accession:L26954
to show the Reference Gene Catalog entries associated with this GenBank nucleotide sequence, regardless of its version number. (The Reference Gene Catalog contains the latest version of a sequence record; if you don't know what version number is the latest, enter only the accession as your query, without any dot or version number.)
-
organism fields:
The whitelisted_taxa and blacklisted_taxa data fields below are used for retrieving organism-specific results. Specifically, they are used to screen for known resistance-causing point mutations within an organism group, and for common, non-informative genes, respectively.
Point mutations are currently identified for 28 bacterial taxonomic groups, which are listed here. Note that rRNA mutations will not be screened if only a protein file is provided. To screen known Shigella mutations, use Escherichia as the organism. See Organism option below for more details.
-
Whitelisted taxa (whitelisted_taxa)
The whitelisted_taxa data field indicates for which taxa this element is curated for mutational resistance mechansims.
An example of a whitelisted sequence is the 16S_A1055G point mutation in E. coli.
See the AMRFinderPlus documentation for a list of taxa where resistance mechanisms based on mutations are curated. Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: whitelisted_taxa:searchterm
- Search for: whitelisted_taxa:Escherichia
to list the resistance-causing point mutations found in the Escherichia taxonomic group (i.e., Escherichia coli and Shigella spp., Escherichia fergusonii).
- The AMRFinderPlus software automatically looks for whitelisted sequences if an organism is specified during a search. For example, if AMRFinderPlus is run with Escherichia in the organism field, then your isolate will be screened for the presence of point mutations that confer antimicrobial resistance in this taxonomic group (such as the 16S_A1055G point mutation). If AMRFinderPlus is run without Escherichia in the organism field, then your isolate will not be screened for the presence of this point mutation.
-
Blacklisted taxa (blacklisted_taxa)
The blacklisted_taxa data field screens for genes that are common within a taxonomic group, and are therefore non-informative with regard to antimicrobial resistance.
An example of a blacklisted sequence is fieF which is blacklisted for both E. coli and Salmonella.
The available values in blacklisted_taxa currently include:- Escherichia > Escherichia coli and Shigella spp., Escherichia fergusonii
- Klebsiella > Klebsiella pneumoniae and Klebsiella oxytoca
- Salmonella > Salmonella enterica
- Staphylococcus > Staphylococcus pseudintermedius
- Vibrio > Vibrio cholerae
Examples:- To search this field directly, enter a query such as: blacklisted_taxa:searchterm
- Search for: blacklisted_taxa:Klebsiella
to list genes that have been blacklisted in the Klebsiella taxonomic group (i.e., Klebsiella pneumoniae and Klebsiella oxytoca). - Search for: blacklisted_taxa:Escherichia AND blacklisted_taxa:Salmonella
to list genes that have been blacklisted in both the Escherichia taxonomic group (i.e., Escherichia coli and Shigella spp., Escherichia fergusonii), and in Salmonella.
- The AMRFinderPlus software automatically excludes blacklisted sequences if an organism is specified during a search. For example, if AMRFinderPlus is run with either Escherichia or Salmonella in the organism field, then your isolate will be screened for the presence of common genes in the taxonomic group, and those common genes will be eliminated from the AMRFinderPlus results. For example, the fieF gene will not be reported even if it is present in your isolate, since fieF is ubiquitous in both of these taxa and reporting it does not provide useful information.
-
Whitelisted taxa (whitelisted_taxa)
-
Curated RefSeq start (curated_refseq_start)
Did curators alter the start coordinate from the GenBank record when making the RefSeq record? The allowable values for this field are Yes or No, and must be written with a leading upper case letter.
A "Yes" indicates that NCBI RefSeq curators either changed the translation start site (in the NG_* genomic sequence record) from what was shown on the corresponding GenBank record, or provided start and stop coordinates that the GenBank record lacked.
The data field name is also case sensitive and should be written in all lower case, as shown in the example below. (Separate sections of this document provides additional details about case sensitive searches and accession prefixes that appear in the Pathogen Detection project.)
Examples:- To search this field directly, enter a query such as: curated_refseq_start:searchterm
- Search for: curated_refseq_start:Yes
to show all genes an alleles that have a curated RefSeq start.
-
GenBank start (genbank_start)
The start coordinate of the reference sequence for this element on the GenBank nucleotide sequence record. This field should always be lower than the GenBank stop field regardless of the GenBank strand
-
GenBank stop (genbank_stop)
The stop coordinate of this reference sequence for this element on the GenBank nucleotide sequence record. This field should always be higher than the GenBank start field regardless of the GenBank strand.
-
GenBank strand (genbank_strand)
The strand (+/-) on which the reference sequence occurs, relative to the nucleotide sequence that appears in the genbank_nucleotide_accession listed for the gene or allele.
-
RefSeq start (refseq_start)
The start coordinate of this reference sequence for this element on the RefSeq nucleotide sequence record. This field should always be lower than the RefSeq stop field regardless of the RefSeq strand
-
RefSeq stop (refseq_stop)
The stop coordinate of this reference sequence for this element on the RefSeq nucleotide sequence record. This field should always be higher than the RefSeq start field regardless of the RefSeq strand.
-
RefSeq strand (refseq_strand)
The strand (+/-) of reference sequence for this element, relative to the nucleotide sequence that appears in the refseq_nucleotide_accession listed for the gene or allele.
-
PubMed reference (pubmed_reference)
Links to references describing gene, if available. The value in the data field is a PubMed identifier (PMID). Clicking on an entry in this field will take you to the page for that paper in PubMed.
-
Synonyms (synonyms)
Other symbols used to refer to this element / gene in the literature.
-
Links (links_count)
Links will contain hyperlinks to other resources. Currently the field contains links to PubChem when there is an identical protein within PubChem for the current protein.
Output from the Pathogen Detection Reference Gene Catalog
- Upon opening the Pathogen Detection Reference Gene Catalog, a table displays data for all genes and alleles that are currently in the catalog.
- Every row in the Pathogen Detection Reference Gene Catalog display is a reference gene or a point mutation.
- The data available for each item can include gene or allele name, product name, type, subtype, class, subclass, and more, as available. (See the Pathogen Detection Reference Gene Catalog data fields for a complete list.) Some of the data elements, such a accessions for corresponding protein and nucleotide sequence records and publications, link to additional information in related databases such as RefSeq, GenBank, and PubMed.
- The genes and point mutations can be sorted by clicking on column headers, faceted by using filters (e.g., class:AMINOGLYCOSIDE), or searched using basic or advanced search techniques.
- Download the list of elements and their metadata shown. Click on the Download button just above the main data table and select File type: Table. From there you can select Tab-delimited (.tsv) or Comma-delimited (.csv) and set the filename. Clicking Download will download the data shown in the table filtered by the search and with the visible columns included. See the Download the Reference Gene Catalog data section for more information and how to download sequences.
- The "Filters" menu options in the Reference Gene Catalog enable you to facet or subset the data in a variety of ways, and therefore can be used to refine your results, whether you have done a basic search or an advanced search.
- By default, each filter displays the top 10 terms (based on the number of genes/alleles retrieved by a term). The "more [+]" option displays up to the top 100 terms, listed alphabetically within that set of top 100.
- Filters are generated on the fly. The choices listed in the "Filters" tab depend on the data set you are currently displaying in the browser, and reflect the attributes of the genes and alleles in that data set.
- A separate section of this document provides additional information about Filters.
- The columns displayed by the Reference Gene Catalog reflect the data fields. By default, the Reference Gene Catalog displays only a subset of the available data fields.
- You can use the "Choose Columns" option at the top of the tabular list of genes in order to remove columns, select additional columns to display, and/or change the order of the columns.
- The options you select will persist within a given browser (e.g., Chrome, Edge, Internet Explorer, Firefox, Safari) until that browser's cookies are cleared/reset.
Use cases/sample searches of the Pathogen Detection Reference Gene Catalog
- Find multidrug resistant genes
- Find carbapenem resistant genes
- Find point mutations in Escherichia that confer resistance to quinolones
- Open the NCBI Pathogen Detection Reference Gene Catalog.
- Open the "Filters" function.
- By default, each filter shows the top 100 terms (based on the number of genes/alleles retrieved by a term).
- In the Class section of the filters, scroll down to find MULTIDRUG or search for MULTIDRUG in the search box.
- "MULTIDRUG" now appears as an option under Class. Select that option. Upon this action, the Filters display will refresh itself to show only the set of filters that apply to that class of antibiotics, and the tabular list of genes and alleles will refresh itself to show only the items that fall in that class.
class:MULTIDRUG
Separate sections of this file provides details about filters, and about how to directly search specific data fields, such as the class and subclass fields, and case sensitive searches.
- Open the NCBI Pathogen Detection Reference Gene Catalog.
- Open the "Filters" function.
- In the Class section of the filters, select "BETA-LACTAM." Upon this action, the Filters display will refresh itself to show only the set of filters that apply to the Beta-Lactam class.
- The Subclass section of the filters will now list "CARBAPENEM" as an option. Check the box for CARBAPENEM to show the genes that confer resistance to that subclass of antibiotics.
subclass:CARBAPENEM
Separate sections of this file provides details about filters, and about how to directly search specific data fields, such as the class and subclass fields, and case sensitive searches.
- Open the NCBI Pathogen Detection Reference Gene Catalog.
- Open the "Filters" function.
- By default, each filter shows the top 100 terms (based on the number of genes/alleles retrieved by a term).
- In the Organism section of the filters, select "Escherichia." Upon this action, the Filters display will refresh itself to show only the set of filters that apply to Escherichia.
- In the Subtype section of the filters, select "POINT." Upon this action, the Filters display will refresh itself to show only the set of filters that apply to Escherichia point mutations.
- In the Subclass section of the filters, scroll to "QUINOLONE" or type that term in the search box. It now appears as an option. Select that option.
- The resulting output is a list of Escherichia point mutations that confer resistance to quinolone antibiotics.
organism:Escherichia AND subtype:POINT AND subclass:QUINOLONE
Separate sections of this file provides details about filters, and about how to directly search specific data fields, such as the organism, type, subtype, class, and subclass fields, and case sensitive searches.
AMRFinderPlus
What is AMRFinderPlus?
AMRFinderPlus - Identifies antimicrobial resistance (AMR) genes and point mutations in assembled nucleotide and protein sequences. AMRFinderPlus also identifies select virulence and stress resistance genes. AMRFinderPlus compares isolate genomes against the reference protein set using BLAST and against the HMM set using HMMER, and uses the gene hierarchy to provide the most specific protein assignment to antimicrobial resistant protein or family, if present in the query set of proteins. The original AMRFinder identifies acquired antimicrobial resistance (AMR) genes, as well as point mutations that confer antimicrobial resistance, in either protein datasets or nucleotide data, including genomic data. AMRFinderPlus identifies the AMR genes and point mutations that are found by the original AMRFinder, plus it identifies select members of additional classes of genes such as virulence factors, biocide, heat, acid, and metal resistance genes. Unlike other AMR gene detection methods that report the best hit, AMRFinderPlus reports the specific gene symbol based on the available evidence. For example, when presented with a novel blaKPC allele that is nearly identical to blaKPC-2, closest hit tools might return blaKPC-2, but AMRFinderPlus would call it as blaKPC (illustrated example). More details about the tool are provided in a publication by Feldgarden M, et al., 2021.
- Install AMRFinderPlus from: https://github.com/ncbi/amr/wiki
- Download data files:
The AMRFinderPlus software uses the following data sets, which can be downloaded from the links below:- Pathogen Detection Reference Gene Catalog:
- The collection of curated genes used in AMRFinderPlus, which includes two subsets of genes/alleles: (1) "Core:" highly curated, AMR-specific genes; (2) "Plus:" biocide and stress resistance, general efflux, virulence, or antigenicity genes.
- Read more about the Reference Gene Catalog
- browse/search the Reference Gene Catalog
- Download: https://ftp.ncbi.nlm.nih.gov/pathogen/Antimicrobial_resistance/AMRFinderPlus/database
- Pathogen Detection HMM Catalog (Hidden Makrov Models):
- A library of Hidden Markov Models (HMMs) that was created, calibrated, and annotated at NCBI, in order to create a hierarchical classification system for AMR proteins, with fine divisions of both recognized families and additional groups to sensitively and accurately identify AMR gene sequences.
- Read more about the Reference HMM Catalog
- browse/search the Reference HMM Catalog
- Download: https://ftp.ncbi.nlm.nih.gov/hmm/NCBIfam-AMRFinder/
- Reference Gene Hierarchy:
- A hierarchical framework of gene families, symbols, and names to provide the most specific protein assignment to antimicrobial resistant protein or family. Both the reference proteins and the HMMs are placed into this hierarchy. Unlike other AMR gene detection methods that report the best hit, AMRFinderPlus reports the specific gene symbol based on the available evidence. For example, when presented with a novel blaKPC allele that is nearly identical to blaKPC-2, closest hit tools might return blaKPC-2, but AMRFinder would not incorrectly call it the blaKPC-2 allele (illustrated example).
- Read more about the Reference Gene Hierarchy
- Browse/search the Reference Gene Hierarchy
- Download: Reference Gene Hierarchy in tab-delimited format
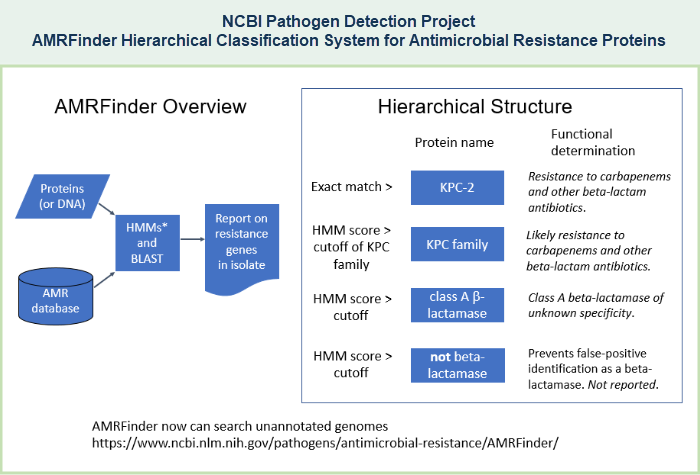
- Pathogen Detection Reference Gene Catalog:
- See the AMRFinderPlus documentation on GitHub for details about intepreting AMRFinderPlus results: https://github.com/ncbi/amr/wiki/Interpreting-results
- Overview: AMRFinderPlus
- AMRFinder (original version) identifies acquired antimicrobial resistance (AMR) genes, as well as point mutations that confer antimicrobial resistance, in either protein datasets or nucleotide data, including genomic data. Additional details are provided in the documentation for AMRFinder in GitHub (https://github.com/ncbi/amr/wiki/v1-Home).
- AMRFinderPlus identifies the AMR genes and point mutations that are found by the original AMRFinder, plus it identifies select members of additional classes of genes such as virulence factors, biocide, heat, acid, and metal resistance genes. Additional details are provided in the documentation for AMRFinderPlus in GitHub (https://github.com/ncbi/amr/wiki).
MicroBIGG-E (Microbial Browser for Identification of Genetic and Genomic Elements)
- What is MicroBIGG-E?
- Where to access MicroBIGG-E?
- Search tips
- Data fields include:
- Isolate data fields, such as:
- Element data fields, such as:
- Reference data fields, such as:
- Analysis results (Element vs Reference) data fields, such as:
- Analysis log data fields, such as:
- see a list of all MicroBIGG-E data fields
- Output
- Use cases/sample searches of MicroBIGG-E
- Identify isolates that have same set of genes and determine if they co-occur on the same contig
- Identify partial gene sequences in the middle of contigs
- Find the isolates and alleles described by a publication of interest
- Display isolates in isolates browser that have the same set of genes co-occurring on the same contig
- Display hits from isolates with co-occurring genes
What is MicroBIGG-E?
- MicroBIGG-E contains genetic and genomic elements identified in assemblies analyzed by AMRFinderPlus as part of the Pathogen Detection Pipeline. See the AMRFinderPlus wiki for more information on how AMRFinderPlus works and the Pathogen Detection Reference Gene Catalog for a list of the elements that AMRFinderPlus is searching for.
- MicroBIGG-E will be updated each time an organism group is updated in the Isolates Browser.
- It contains the genetic and genomic elements that have been found in isolate genomes that have been published in GenBank. (This is in contrast to the Isolates Browser, which contains isolates that have been published in GenBank as well as those awaiting submission to GenBank.) The output is the results of AMRFinderPlus analyses, as described in the data processing pipeline section of this document.
- The MicroBIGG-E will initially include genes, alleles, and point mutations.
- Every row in the MicroBIGG-E display is an anti-microbial resistance (AMR), stress response, and/or virulence gene that has been identified in an isolate assembly by the data processing pipeline, with information about the method used to identify it, supporting evidence, and the element's type, subtype, class, subclass, and more.
- The purpose of MicroBIGG-E is to enable researchers to obtain detailed information about the element as well as the actual contigs that contain a genetic or genomic element of interest, in order to conduct further analysis.
- The Pathogen Detection pipeline uses two assemblers, a de novo assembler and a targeted assembler (SAUTE) to increase assembly sensitivity and accuracy for AMR genes. A region of the genome may therefor appear in two contigs so it looks like there are duplicated genes. For this reason the copy number for AMR genes in MicroBIGG-E will often be higher than appears in the actual isolate.
- MicroBIGG-E and the Pathogens Isolates Browser are related resources and are integrated with each other.
- The main similiarities between the resources are their shared search engine and similar search techniques:
- Both use the SOLR query language and allow searches by a wide variety of text terms.
- The search tips provided in the Isolates Browser help documentation therefore also apply to MicroBIGG-E, such as basic search techniques, advanced search techniques, case sensitive versus case insensitive searches, and the availability of "filters" to refine search results.
- The main differences between the resources are the scope of data being searched, the set of data fields (and filters, which are based on data fields) that are available for searching, and the columns that are shown in the display of search results: (The MicroBIGG-E data fields are described below, and the Reference Gene Catalog data fields and Pathogens Isolates Browser data fields are described in the help for each of those resources.)
- The Pathogens Isolates Browser searches all isolate genomes in the Pathogen Detection project which have been deposited in GenBank, as well as corresponding metadata that are described under types of data.
- Every row in the Isolates Browser is an assembled isolate, possibly with antimicrobial resistance (AMR), virulence, and/or stress response genotype data, and antibiotic susceptibility (AST) phenotype data, as available.
- The Isolates Browser help documentation describes the available data fields and output.
- The MicroBIGG-E searches the collection of genetic and genomic elements, such as genes related to antimicrobial resistance, stress resistance, and virulence, that have been identified in the isolates through the data processing pipeline.
- Every row in the MicroBIGG-E display is an anti-microbial resistance (AMR), stress response, and/or virulence gene that has been identified in an isolate by the data processing pipeline, with information about the method used to identify it, supporting evidence, and the element's type, subtype, class, subclass.
- The MicroBIGG-E data fields are described below, and are reflected in the columns displayed in the MicroBIGG-E output.
- The Pathogens Isolates Browser searches all isolate genomes in the Pathogen Detection project which have been deposited in GenBank, as well as corresponding metadata that are described under types of data.
- The Pathogens Isolates Browser and MicroBIGG-E are linked using Cross-browser selection.
Where to access MicroBIGG-E
and the raw data behind it is available at Google Cloud. You can also access MicroBIGG-E directly from the links below:
Browse/Search MicroBIGG-E:
/pathogens/isolates#/microbigge/.
Download the MicroBIGG-E data:
Click the "Download" button in the header of the MicroBIGG-E table to download data. You can either download a tab-delimited or csv formatted representation of the table view or a set of sequences under the "Dataset" selection.
- Table downloads can be in either Tab-delimited (.tsv) format or Excel comma-delimited format (.csv), and have a maximum of 100,000 rows.
- Datasets downloads contain protein or nucleotide data related to the elements shown in the table. These can be the DNA sequence of the elements, the elements plus flanks (up to 2,000 bp), the entire contig containing the elements (max 1,000 contigs), or the amino-acid sequences of the protein elements.
 GCP BigQuery Full table access using SQL. See MicroBIGG-E data at Google Cloud Platform for more information on how to get full MicroBIGG-E data on Google Cloud in BigQuery.
GCP BigQuery Full table access using SQL. See MicroBIGG-E data at Google Cloud Platform for more information on how to get full MicroBIGG-E data on Google Cloud in BigQuery.
Search tips for MicroBIGG-E
- MicroBIGG-E can be searched by the terms that appear in any of the data fields described below. A search example is provided after each data field description, when possible.
- The query tips described in the Isolates Browser help > basic search section also apply to MicroBIGG-E, such as searches for multiple terms, special characters, phrase searches, case sensitive vs. case insensitive searches, etc.
- The query tips described in the Isolates Browser help > advanced search section also apply to the MicroBIGG-E, because both resources use the SOLR query language.
- The main difference is the data fields that are available to be searched, because each resource has its own set of data fields. (The data fields in MicroBIGG-E are the same as the data fields in the Pathogen Detection Reference Gene Catalog, which are different from the data fields in the Isolates Browser.)
- The query tips described in the Isolates Browser help also apply to MicroBIGG-E, such as searches for multiple terms, special characters, phrase searches, case sensitive vs. case insensitive searches, etc.
- The "Filters" menu options in the MicroBIGG-E enable you to facet or subset the data in a variety of ways, and therefore can be used to refine your results, whether you have done a basic search or an advanced search.
- Each filter displays counts of elements next to each term in the filter. Note that these counts are for elements in the browser, and may not accurately describe the number of genes in actual isolates because Pathogen Detection assemblies use both de novo and guided assemblies which may represent the same gene in an assembly multiple times.
- By default, each filter displays the top 100 terms (based on the number of isolates retrieved by a term) listed by count of value within that set of top 100. Use the search box to search for filters not in the top 100. Note that:
- A Boolean "OR" is applied if multiple items are checked in the same filter field. This way you can choose multiple values in the same filter. For example:
- Open the "Filters" tab of the MicroBIGG-E, then check the boxes for "Stress" and for "Virulence" in the "Type" filter. The system will retrieve genetic/genomic elements that are associated with either stress resistance or with virulence.
- A Boolean "AND" is applied if you select items in several different filter fields (Type, Class, etc). For example:
- Open the "Filters" tab of the MicroBIGG-E web interface, then check the boxes for "Point" in the "Subtype" filter and "Quinolone" in the "Class" filter. The system will retrieve genetic/genomic elements that meet both of your specified criteria (in this case, point mutations that confer resistance to quinolones).
- A Boolean "OR" is applied if multiple items are checked in the same filter field. This way you can choose multiple values in the same filter. For example:
- As explained in the Isolates Browser help, Filters are generated on the fly. As a result, the terms that are listed under each filter will depend on the data set you are currently displaying in the browser. That is also true for the filters in the MicroBIGG-E.
Data Fields in MicroBIGG-E
Each data field reflects an available column in the MicroBIGG-E web interface. The output section of this document provides tips on how to customize the display, using the "choose columns" function.
Please note: in the list of available data fields below:
- The term shown in the regular font is the display name (column header) shown by the MicroBIGG-E web interface. The term shown in (italics) is the name of the corresponding data field, if you want to search that field directly.
- For example, one data field is listed as: Method (amr_method). The term "Method" appears in the MicroBIGG-E column header, and "amr_method" (with an underscore bar instead of a space) is the string you should use if you want to search that data field directly.
- Brief italicized search examples are also provided for each data field, when possible, showing how to query the data field directly. The values represent text strings exactly as they appear in the data fields, including upper case and lower case letters, special characters such as hyphens, etc. The data field names and values are case sensitive.
Note that each field is written in this format: Display name (data_field_name)
The "Display name" is the column header that appears in the MicroBIGG-E web interface, and the "data_field_name" is the case-sensitive string you should enter if you want to search the data field directly using a SOLR query:
Isolate data fields:
-
Scientific name (scientific_name)
This data field also appears in the Pathogens Isolates Browser; a description of Scientific name and examples of queries for that field appear in the Isolates Browser data fields help section. -
Organism group (taxgroup_name)
This data field also appears in the Pathogens Isolates Browser; a description of Organism group and examples of queries for that field appear in the Isolates Browser data fields help section. -
Serovar (serovar)
This data field also appears in the Pathogens Isolates Browser; a description of Serovar and examples of queries for that field appear in the Isolates Browser data fields help section. -
Strain (strain)
This data field also appears in the Pathogens Isolates Browser; a description of Strain and examples of queries for that field appear in the Isolates Browser data fields help section. -
Isolate (target_acc)
This data field also appears in the Pathogens Isolates Browser; a description of Isolate and examples of queries for that field appear in the Isolates Browser data fields help section. -
Isolation source (isolation_source)
This data field also appears in the Pathogens Isolates Browser; a description of Isolation Source and examples of queries for that field appear in the Isolates Browser data fields help section. -
Isolation type (epi_type)
This data field also appears in the Pathogens Isolates Browser; a description of Isolation type and examples of queries for that field appear in the Isolates Browser data fields help section. -
BioSample (biosample_acc)
This data field also appears in the Pathogens Isolates Browser; a description of BioSample and examples of queries for that field appear in the Isolates Browser data fields help section. -
Collection date (collection-date)
This data field also appears in the Pathogens Isolates Browser; a description of Collection Date and examples of queries for that field appear in the Isolates Browser data fields help section. -
Location (geo_loc_name)
This data field also appears in the Pathogens Isolates Browser; a description of Location and examples of queries for that field appear in the Isolates Browser data fields help section. -
Host (host)
This data field also appears in the Pathogens Isolates Browser; a description of Host and examples of queries for that field appear in the Isolates Browser data fields help section. -
Run (Run)
This data field also appears in the Pathogens Isolates Browser; a description of Run and examples of queries for that field appear in the Isolates Browser data fields help section. -
Assembly (asm_acc)
This data field also appears in the Pathogens Isolates Browser; a description of Assembly and examples of queries for that field appear in the Isolates Browser data fields help section.
-
Element symbol (element_symbol)
The symbol assigned to the element by AMRFinderPlus. Examples include an allele symbol (blaKPC-2), a protein symbol (blaKPC), or a point mutation symbol (gyrA_G81D). It can also be a very broad symbol representing a large family of proteins (bla) that you would not find in the reference gene catalog. This happens when AMRFinderPlus lacks evidence to use a more specific element symbol.
Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: element_symbol:searchterm
- Search for: element_symbol:blaKPC
to show all genetic/genomic elements with that exact symbol. - Search for: element_symbol:blaKPC OR element_symbol:blaKPC-2
to show all genetic/genomic elements that have either of those exact symbols.
-
Element name (element_name)
The name of the element assigned by AMRFinderPlus.
Data field names and values are case sensitive, as shown in the examples below. Use quotes to search for phrases, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: element_name:searchterm
- Search for: element_name:"KPC family carbapenem-hydrolyzing class A beta-lactamase"
to show genetic/genomic elements with that name. - Search for: element_name:"KPC family carbapenem-hydrolyzing class A beta-lactamase" OR element_name:"carbapenem-hydrolyzing class A beta-lactamase KPC-2"
to show all genetic/genomic elements that have either of those names.
-
Element length (element_length)
The length of this element in amino acids (AA) for protein elements, and in base pairs (bp) for nucleotide elements.
Data field names and values are case sensitive, as shown in the examples below.
This data field can be queried by a range search, as shown in the example below.
Examples:- To search this field directly, enter a query such as: element_length:searchterm
- To search for a range of values, enter a query such as: element_length:[value1 TO value2]
- Search for: element_length:234
to show genetic/genomic elements are have a length of 234 amino acids (or 234 nucleotides). - Search for: element_length:[200 TO 250]
to show genetic/genomic elements that range in length between 200 and 250 amino acids (or between 200 and 250 nucleotides).
-
Protein (protein_acc)
The accession of the protein sequence record for this element.
Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: protein_acc:searchterm
- Search for: protein_acc:WP_004199234.1
to show genetic/genomic elements that have the protein sequence shown in the RefSeq record WP_004199234.1. This search retrieves more genetic/genomic elements from a large number of isolates, because the sequence has been found to be a multipecies protein. - Search for: protein_acc:WP_124042569.1
to show the genetic/genomic elements that have the protein sequence shown in the RefSeq record WP_124042569.1. As of May 23, 2020, this search retrieves a single element, from the E. coli isolate PDT000411318.1.
-
Contig (contig_acc)
The accession of the contig sequence record on which this element appears.
Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: contig_acc:searchterm
- Search for: contig_acc:NZ_UWVC01000003.1
to show the genetic/genomic elements that have been identified on the sequence of contig NZ_UWVC01000003.1.
-
Start (start_on_contig)
The start coordinate for the element on the contig sequence.
-
Stop (end_on_contig)
The stop coordinate for the element on the contig sequence.
-
Strand (strand)
The strand (+/-) on which the genetic or genomic element appears, relative to the nucleotide sequence that appears in the contig accession listed for the element. -
Type (type)
Classification for the type of gene found, such as AMR, STRESS, or VIRULENCE.
A more detailed description of the type and subtype fields is available on the AMRFinderPlus wiki
This data field also appears in the Pathogen Detection Reference Gene Catalog; a description of Type and examples of queries for that field appear in the Reference Gene Catalog data fields help section.
(In general, type and subtype refer to the category of gene or genetic element, while class and subclass refer to the substrate.) -
Subtype (subtype)
Classification for the subtype of gene found. A more detailed description of the type and subtype fields is available on the AMRFinderPlus wiki
This data field also appears in the Pathogen Detection Reference Gene Catalog; a description of Subtype and examples of queries for that field appear in the Reference Gene Catalog data fields help section.
(In general, type and subtype refer to the category of gene or genetic element, while class and subclass refer to the substrate.) -
Class (class)
Class of resistance for "core" genes (see scope), and typing information for some virulence genes.
This data field also appears in the Pathogen Detection Reference Gene Catalog; a description of Class and examples of queries for that field appear in the Reference Gene Catalog data fields help section.
(In general, type and subtype refer to the category of gene or genetic element, while class and subclass refer to the substrate.) -
Subclass (subclass)
Where it is known, "Subclass" provides a more specific definition of the particular antibiotics or classes that are affected by the gene or point mutation (e.g., that are resisted by the gene/allele). While most subclass designations are self-explanatory, a few others have particular meanings. Specifically, "CEPHALOSPORIN" is equivalent to the Lahey 2be definition; "CARBAPENEM" means the protein has carbapenemase activity, but it might or might not confer resistance to other beta-lactams; "QUARTERNARY AMMONIUM" are quarternary ammonium compounds. In addition, stx subtypes (e.g., STX2E) and intimin subtypes (e.g., ALPHA) are defined for Shiga toxin proteins (class of STX1 or STX2) and intimins (class of INTIMIN) respectively. Where the phenotypic information is incomplete, contradictory, or unclear, the "Class" value is used for the "Subclass" value.
More information about the class and subclass fields can be found on the AMRFinderPlus wiki
This data field also appears in the Pathogen Detection Reference Gene Catalog; a description of Subclass and examples of queries for that field appear in the Reference Gene Catalog data fields help section.
(In general, type and subtype refer to the category of gene or genetic element, while class and subclass refer to the substrate.) -
Scope (scope)
This data field also appears in the Pathogen Detection Reference Gene Catalog; a description of Scope and examples of queries for that field appear in the Reference Gene Catalog data fields help section.
-
Closest reference accession (closest_reference_acc)
The accession of closest reference sequence. Note that only one reference will be chosen if the blast hit is equidistant from multiple references (NA if HMM-only hit). For point mutations the reference is the sensitive "wild-type" allele, and the element symbol describes the specific mutation. Check the Reference Gene Catalog for more information on specific mutations or reference genes.
Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: closest_reference_acc:searchterm
- Search for: closest_reference_acc:WP_001083725.1
to show genetic/genomic elements whose protein sequence is most closely related to the sequence in RefSeq record https://www.ncbi.nlm.nih.gov/protein/WP_001083725.1.
Note that some elements retrieved by the search above will list that accession in both the closest_reference_acc and protein_acc columns, while other proteins will list it only in the closest_reference_acc column. You can retrieve either subset with the following searches:
Search for: closest_reference_acc:WP_001083725.1 AND protein_acc:WP_001083725.1
Search for: closest_reference_acc:WP_001083725.1 NOT protein_acc:WP_001083725.1
-
Closest reference name (closest_reference_name)
The name of closest reference sequence.
Data field names and values are case sensitive, as shown in the examples below. Use quotes to search for phrases, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: closest_reference_name:searchterm
- Search for: closest_reference_name:"trimethoprim-resistant dihydrofolate reductase DfrA12"
to show genetic/genomic elements whose closes RefSeq protein sequence is named "trimethoprim-resistant dihydrofolate reductase DfrA12."
-
Reference element length (reference_element_length)
Length of the reference sequence in amino acids (AA) for protein elements, and in base pairs (bp) for nucleotide elements.
Data field names and values are case sensitive, as shown in the examples below.
This data field can be queried by a range search, as shown in the example below.
Examples:- To search this field directly, enter a query such as: reference_element_length:searchterm
- To search for a range of values, enter a query such as: reference_element_length:[value1 TO value2]
- Search for: reference_element_length:284
to show genetic/genomic elements whose reference elements have a length of 234 amino acids (or 234 nucleotides). - Search for: reference_element_length:[200 TO 250]
to show genetic/genomic elements whose reference elements range in length between 200 and 250 amino acids (or between 200 and 250 nucleotides).
-
HMM Accession (hmm_acc)
The accession of the Hidden Markov Model (HMM) that hits this element above cutoff (if any). Clicking the HMM accession will take you to the HMM page in the Protein Family Models database. From that page you can download the HMM itself and get additional information including the curated cutoffs, the seed alignment, and RefSeq sequences identified by this HMM.
Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: hmm_acc:searchterm
- Search for: hmm_acc:NF000053.2
to show genetic/genomic elements that have a match to the Hidden Markov Model with accession NF000053.2 (trimethoprim-resistant dihydrofolate reductase DfrA12).
-
HMM Description (hmm_description)
The name of the Hidden Markov Model (HMM) that hits this element (if any).
Data field names and values are case sensitive, as shown in the examples below. Use quotes to search for phrases, as shown in the example below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: hmm_description:searchterm
- Search for: hmm_description:"trimethoprim-resistant dihydrofolate reductase DfrA12"
to show genetic/genomic elements that have a match to the Hidden Markov Model with the name "trimethoprim-resistant dihydrofolate reductase DfrA12."
-
Method (amr_method)
The method used by AMRFinderPlus to identify this element. A separate section of this document provides a table that summarizes AMRFinderPlus methods that are used by the tool to analyze pathogen isolate genome assemblies and identify genetic and genomic elements. The AMRFinderPlus Wiki provides additional details about the methods.
Data field names and values are case sensitive, as shown in the examples below. Additional sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards (such as the asterisk or question mark).
Examples:- To search this field directly, enter a query such as: amr_method:searchterm
- Search for: amr_method:HMM
to show proteins that were found by HMM only, more distant to reference proteins than our BLAST cutoffs. - Search for: amr_method:POINTN OR amr_method:POINTP OR amr_method:POINTX
to show point mutations that were identified using nucleotide BLAST (BLASTN), protein BLAST (BLASTP), or translated BLAST (BLASTX).
-
Alignment length (align_length)
The length of the alignment between the genetic/genomic element, which was identified by AMRFinderPlus in the isolate genome assembly, and the reference element. The length is measured in amino acids (AA) for protein elements, and in base pairs (bp) for nucleotide elements.
Data field names and values are case sensitive, as shown in the examples below.
This data field can be queried by a range search, as shown in the example below.
Examples:- To search this field directly, enter a query such as: align_length:searchterm
- To search for a range of values, enter a query such as: align_length:[value1 TO value2]
- Search for: align_length:[200 TO 250]
to show genetic/genomic elements whose alignment to the closest reference element ranges in length between 200 and 250 amino acids (or between 200 and 250 nucleotides).
-
% Identity (pct_ref_identity)
The percent of identical amino acids or base pairs within the aligned region of the genetic/genomic element (identified by AMRFinderPlus in the isolate genome assembly) and the reference element.
Data field names and values are case sensitive, as shown in the examples below.
This data field can be queried by a range search, as shown in the example below.
Examples:- To search this field directly, enter a query such as: pct_ref_identity:searchterm
- To search for a range of values, enter a query such as: pct_ref_identity:[value1 TO value2]
- Search for: pct_ref_identity:100
to show genetic/genomic elements that have a 100% identity to the reference element, within the aligned region. - Search for: pct_ref_identity:[98 TO 100]
to show genetic/genomic elements that have an identity that ranges from 98% to 100% to the reference element, within the aligned region.
-
% Coverage (pct_ref_coverage)
The proportion of the reference sequence covered by the alignment between the target element and the reference element.
For example, a coverage of 90% means that the alignment between the target element and the reference element covers 90% of the reference sequence's length.
Data field names and values are case sensitive, as shown in the examples below.
This data field can be queried by a range search, as shown in the example below.
Examples:- To search this field directly, enter a query such as: pct_ref_coverage:searchterm
- To search for a range of values, enter a query such as: pct_ref_coverage:[value1 TO value2]
- Search for: pct_ref_coverage:100
to show genetic/genomic elements whose alignment to the closest reference element covers 100% of the reference element's length. - Search for: pct_ref_coverage:[50 TO 75]
to show genetic/genomic elements whose alignment to the closest reference element covers 50% to 75% of the reference element's length.
-
Contig coverage (contig_coverage)
Contig coverage is the mean coverage of aligned reads for the contig containing this hit. This is a decimal (floating point) number > 0, not a percentage.
Data field names and values are case sensitive, as shown in the examples below.
This data field can be queried by a range search, as shown in the example below.
Example:- To search this field directly, enter a query such as: contig_coverage:searchterm
- To search for a range of values, enter a query such as: contig_coverage:[value1 TO value2]
- Search for: contig_coverage:[96 TO 106]
to show genetic/genomic elements that have a contig coverage between 96 and 106.
-
Relative assembly coverage (rel_asm_cov)
This is the mean coverage by aligned reads of the entire contig divided by the mean coverage by aligned reads of the entire assembly. Mathematically the value is contig_coverage / asm_coverage. This is a ratio, a decimal (floating point) number > 0, not a percentage.
Data field names and values are case sensitive, as shown in the examples below.
This data field can be queried by a range search, as shown in the example below.
Example:- To search this field directly, enter a query such as: rel_asm_cov:searchterm
- To search for a range of values, enter a query such as: rel_asm_cov:[value1 TO value2]
- Search for: rel_asm_cov:[1.190 TO 1.202]
to show genetic/genomic elements that have relative assembly coverage between 1.190 TO 1.202. - Search for: rel_asm_cov:[1.1 TO 1.2]
to show genetic/genomic elements that have relative assembly coverage between 1.100 TO 1.200.
-
Assembly coverage (asm_coverage)
Assembly coverage is the mean coverage of aligned reads for the entire assembly. This is a decimal (floating point) number > 0, not a percentage.
Data field names and values are case sensitive, as shown in the examples below.
This data field can be queried by a range search, as shown in the example below.
Example:- To search this field directly, enter a query such as: rel_asm_cov:searchterm
- To search for a range of values, enter a query such as: asm_coverage:[value1 TO value2]
- Search for: asm_coverage:[98 TO 110]
to show genetic/genomic elements that have assembly coverage between 98 TO 110.
-
AMRFinderPlus analysis type (amrfinderplus_analysis_type)
This data field also appears in the Pathogens Isolates Browser; a description of AMRFinderPlus analysis type and examples of queries for that field appear in the Isolates Browser data fields help section. -
AMRFinderPlus version (amrfinderplus_version)
This data field also appears in the Pathogens Isolates Browser; a description of AMRFinderPlus version and examples of queries for that field appear in the Isolates Browser data fields help section. -
PD Ref Gene Catalog Version (refgene_db_version)
This data field also appears in the Pathogens Isolates Browser; a description of PD Ref Gene Catalog Version and examples of queries for that field appear in the Isolates Browser data fields help section.
Output from MicroBIGG-E
- Upon opening the MicroBIGG-E web interface, a table displays data for all genetic and genomic elements that have been identified in isolates genomes that have been deposited into GenBank.
- Every row in the MicroBIGG-E display is an anti-microbial resistance (AMR), stress response, and/or virulence gene that has been identified in an isolate by the data processing pipeline.
- The data available for each item can include gene name, type, subtype, class, subclass, method used to identify the element, supporting evidence, and more, as available. (See the MicroBIGG-E data fields for a complete list.) Some of the data elements, such as accessions for BioSample, nucleotide sequence, and protein sequence records, link to additional information in the corresponding databases.
- The genes can be sorted by clicking on column headers, faceted by using filters (e.g., class:AMINOGLYCOSIDE), or searched using basic or advanced search techniques.
- The "Filters" menu options in the MicroBIGG-E web interface enable you to facet or subset the data in a variety of ways, and therefore can be used to refine your results, whether you have done a basic search or an advanced search.
- By default, each filter displays the top 100 terms (based on the number of genes/alleles retrieved by a term) listed by count of value within that set of top 100.
- Filters are generated on the fly. The choices listed in the "Filters" tab depend on the data set you are currently displaying in the browser, and reflect the attributes of the genes and alleles in that data set.
- A separate section of this document provides additional information about Filters.
- The columns displayed by MicroBIGG-E reflect the data fields. By default, the MicroBIGG-E displays only a subset of the available data fields.
- You can use the "Choose Columns" option at the top of the tabular list of genes in order to remove columns, select additional columns to display, and/or change the order of the columns.
- The options you select will persist within a given browser (e.g., Chrome, Edge, Internet Explorer, Firefox, Safari) until that browser's cookies are cleared/reset.
- It is possible to view isolates that you have identified in MicroBIGG-E in the Isolates Browser
- Click the Cross-browser selection button to the right of the Download button (you must be logged into your myNCBI account for this functionality). By default, all of the isolates for every row of your MicroBIGG-E search will be selected, as indicated by the checkbox column; however, you can deselect rows manually.
- Then click the Show in Isolates button. A new tab will open with the Isolates browser results for the selected elements in MicroBIGG-E. You can then examine clusters of interest using the SNP Tree Viewer, or perform other tasks in the Isolates Browser.
Use cases/sample searches of MicroBIGG-E
- Identify hits from isolates with specific genes that co-occur on the same contig
- Identify partial gene sequences in the middle of contigs
- Find the isolates and alleles described by a publication of interest
- Display isolates in isolates browser that have the same set of genes co-occurring on the same contig
- Display hits from isolates with co-occurring genes
- Open the MicroBIGG-E: Microbial Browser for Identification of Genetic and Genomic Elements.
- Search for contigs with genes of interest (e.g., blaTEM-1 and blaKPC*)
- To do this, enter a search such as:
genes_on_contig:blaTEM-1 AND genes_on_contig:blaKPC*
(Note that field-specified searches are case-sensitive, and separate sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards such as the asterisk.) - Examine if genes of interest co-occur on same contig, either by clicking Download or visual inspection.
- Open the MicroBIGG-E: Microbial Browser for Identification of Genetic and Genomic Elements.
- For a given isolate and gene sequence (i.e., row), exclude columns where method equals "PARTIAL_CONTIG_ENDP" or "PARTIAL_CONTIG_ENDX".
- To do this, enter a search such as:
amr_method:PARTIAL* AND NOT amr_method:PARTIAL_CONTIG_END*
(Note that field-specified searches are case-sensitive, and separate sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards such as the asterisk.)
- Open the MicroBIGG-E: Microbial Browser for Identification of Genetic and Genomic Elements.
- Search for contigs that have a blaKPC gene and a blaTEM-1 allele.
- To do this, enter the following search:
genes_on_contig:blaTEM-1 AND genes_on_contig:blaKPC*
(Note that field-specified searches are case-sensitive, and separate sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards such as the asterisk.) - Click the Cross-browser selection button to the right of the Download button (you must be logged into your myNCBI account for this functionality).
- A new tab will open in the Isolates Browser containing all of the isolates meeting your search criteria. You can then examine clusters of interest using the SNP Tree Viewer, or perform other tasks in the Isolates Browser.
- Open the MicroBIGG-E: Microbial Browser for Identification of Genetic and Genomic Elements.
- Search for hits from isolates that have a blaKPC gene and a blaTEM-1 allele.
- To do this, enter the following search:
genes_on_isolate:blaTEM-1 AND genes_on_isolate:blaKPC*
(Note that field-specified searches are case-sensitive, and separate sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards such as the asterisk.) - Click the Cross-browser selection button to the right of the Download button (you must be logged into your myNCBI account for this functionality).
- A new tab will open in the Isolates Browser containing all of the isolates meeting your search criteria. You can then examine clusters of interest using the SNP Tree Viewer, or perform other tasks in the Isolates Browser.
- Identify the isolates, "Surv196" and "ENT630," from the paper that have these blaACT variants.
- Open the MicroBIGG-E: Microbial Browser for Identification of Genetic and Genomic Elements.
- Search for the isolates in the strain data field by entering the following query:
strain:Surv196 OR strain:ENT630
(Note that field-specified searches are case-sensitive, and separate sections of this document provide tips about search terms that contain special characters (such as the parentheses, hyphens, and apostrophes), and the use of wildcards such as the asterisk.) - Identify the blaACT alleles among the genetic/genomic elements that are retrieved by MicroBIGG-E.
- For the allele of interest, retrieve the corresponding WP_* accession from the Protein database to view the RefSeq protein sequence record. In this case, the accessions for the blaACT proteins that were identified on the isolate genomes are: WP_154123408.1 (on the Surv196 isolate) and WP_152819218.1 (on the ENT630 isolate).
- Note that a WP_* accession can appear in the protein_acc column and/or the closest_reference_acc column.
- Use the choose columns function to display the desired data fields, as only a subset are displayed by default.
- The protein_acc column and closest_reference_acc column might contain the same value (if the protein sequence of the element that was annotated on the isolate genome is identical to the reference protein sequence), or different values (if the protein sequence of the element that was annotated on the isolate genome is not identical to the reference protein sequence).
- If a WP_* accession is not linked to the Protein database, you can search for the accession number directly in the Protein database.
Submit sequence and phenotype data related to AMR
- Submit data for real-time analysis
- Submit antibiograms to the BioSample database
- Request new alleles for Beta-Lactamase, MCR, and Qnr Genes
Download AMR Raw Data
(An overview of the Pathogens FTP site is provided below.)
FTP Site help
What data are on the Pathogens FTP site?
How are the Pathogens data organized on the FTP site?
- Results directory:
Individual phylogenetic trees for each SNP cluster are available in the Pathogens FTP "Results" directory.
Note: Individual phylogenetic trees for each SNP cluster are also accessible from the NCBI Pathogen Detection Isolates Browser. In the Isolates Browser, isolates that have "PDS*" accession number in the "SNP Cluster" column have a link to the SNP Tree Viewer, which provides an interactive display of the SNP cluster. (read more...)
Each folder in this directory contains the data analysis results, such as phylogenetic distance trees, for a given organism group. The folders contain the results of the most current data analyses, as well as archival results from previous analyses. The results for a given organism group are updated daily for each taxgroup, only if new data arrives. Archived results are stored according to the data retention policy.
- Organism group folders - These folders contains the results of data analyses, such as phylogenetic distance trees, that were done on the genome assemblies of isolates within each organism group in the Pathogen Detection Project.
Within an given organism group, the subfolder named with the most recent Pathogen Detection Group accession.version number (PDGxxxxxxxxxx.xxx*) contains the most recent results. The results for a given organism group are updated daily for each taxgroup, only if new data arrives. The "latest_kmer" and "latest_snps" links provide updated links to the most recent results for kmer and SNP analyses, respectively, which may be asynchronously produced (may point to different PDG versions); otherwise they will both point to the most recent PDG version.
- Rapid_reports for select organisms - This directory is a pilot phase test of rapid reporting based solely on wgMLST allele differences and is only operational for a few submitters for a few organisms. The FTP Rapid Reports for a given organism are updated on average within an hour of receiving sequence read submissions for a new isolate.
- Organism group folders - These folders contains the results of data analyses, such as phylogenetic distance trees, that were done on the genome assemblies of isolates within each organism group in the Pathogen Detection Project.
Within an given organism group, the subfolder named with the most recent Pathogen Detection Group accession.version number (PDGxxxxxxxxxx.xxx*) contains the most recent results. The results for a given organism group are updated daily for each taxgroup, only if new data arrives. The "latest_kmer" and "latest_snps" links provide updated links to the most recent results for kmer and SNP analyses, respectively, which may be asynchronously produced (may point to different PDG versions); otherwise they will both point to the most recent PDG version.
- Reference directory:
This directory does NOT include real-time analysis results, and is only based on genomes available in GenBank that are not submitted as part of surveillance networks to SRA. - Antimicrobial_resistance directory:
This directory contains the reference table for AMR genes, and the data files used for AMRFinderPlus.
For more information on NCBI's efforts on antimicrobial resistance, see this page:
/pathogens/antimicrobial-resistance/.
For more information on AMRFinderPlus see this page:
/pathogens/antimicrobial-resistance/AMRFinder/. - Other directories:
For descriptions of the other subdirectories see the FTP README file.
FTP Readme File
- For more information on directory structure, filetypes, and details on what data are available, please see the Pathogens Detection Project FTP readme file available at: https://ftp.ncbi.nlm.nih.gov/pathogen/ReadMe.txt
Data Submissions
Data Processing Pipeline
Overview
NCBI has developed a multi-stage pipeline with two goals: 1) clustering closely related pathogen isolates and 2) identifying antimicrobial resistance genes/proteins in pathogen genomes. The pipeline first assembles the short read sequence data for an isolate into a genome sequence. This includes targeted assembly for certain genes of interest (such as AMR genes) for increased sensitivity. Second, the pipeline clusters the genomes from the assembly process along with the genomes found in GenBank for each organism (see Organism Table for current list). Third, phylogenetic trees are reconstructed after SNP calling within each cluster. The fourth step involves annotation and identification of AMR genes. Details on the full pipeline will be published at a later date. Note: there is a small pilot project pipeline that simply assembles and using the wgMLST scheme to generate a table of nearest neighbors. That pipeline currently only runs for Listeria and Salmonella.I. Assembly pipeline.
- The assembly pipeline uses SKESA to generate de novo assemblies as well as the guided assembler SAUTE to sensitively and comprehensively catalog antimicrobial resistance genes. The current pipeline only assembles Illumina data, assemblies from other sequencing technologies are included when uploaded to GenBank. Note that the de novo and guided assembler pipelines may both independently assemble the same region of the genome, so there will often be duplicated sequence in the final assembly.
II. Clustering.
There are also two different clustering pipelines in operation. Clustering automatically starts once a day for each organism only if new data are submitted.
- The first uses a reference wgMLST scheme (one for each organism if one exists), identifies the loci and alleles in each assembled genome, and uses a 25-allele cut-off to cluster related isolates. This system is gradually being rolled out. Most of the taxgroups with large numbers of isolates submitted are using the wgMLST method. A hard cut-off of 1K isolates is in place before a reference wgMLST scheme is developed, therefore not all organisms will be switched to this system.
- The second uses k-mer distances to first cluster related isolates, then a first pass SNP analysis. Clusters are created using 50-SNP single-linkage clustering. This system is gradually being replaced by the wgMLST but will remain for those organisms that have less than 1K isolates.
III. Phylogenetic tree reconstruction.
For each cluster, a phylogenetic tree is reconstructed from the SNPs for that cluster by using the maximum compatibility criteria.IV. Annotation and antimicrobial gene/protein identification.
Each assembled genome that passes validation criteria will end up in the NCBI Pathogen Detection Isolates Browser. Each SNP cluster is also available, both on FTP as well as in the NCBI Pathogen Detection Isolates Browser. AMR results are available both on FTP and in the browser as a separate column. Rapid Reports are only available on FTP.
New isolates are analyzed using the latest version of the AMRFinderPlus software and the latest version of Pathogen Detection Reference Gene Catalog (read about the Reference Gene Catalog). Older isolates may have been analyzed with earlier versions of the AMRFinderPlus software and the Reference Gene Catalog. There might be occasional updates to annotation on all isolates in special circumstances, such as the identification of a new genes (e.g., mobilized colistin resistance (mcr) genes). Data fields in the Isolates Browser indicate the analysis type (amrfinderplus_analysis_type), AMRFinderPlus version (amrfinderplus_version), and Reference Gene Catalog version (refgene_db_version) that were used in the analysis of a given isolate.
(Separate sections of this file provide Isolates Browser help documentation and an overview of the data available on the FTP site. The AMRFinderPlus wiki provides details about installing and running the program, interpreting the results, and methods used for isolate genome analysis.)
Genotype Categories
The genes identified in an isolate's genome by the NCBI Pathogen Detection data processing pipeline are grouped into genotype categories.The stand-alone AMRFinderPlus software produces a detailed categorization, based on the method used to identify the genotypes. (The AMRFinderPlus wiki provides details about the methods, under "Running AMRFinderPlus > Output Format > Fields > Method".)
The Isolates Browser web interface displays a simplified categorization of genotypes. (The genotype categories appear when you use the choose columns function to display data such as AMR genotypes (AMR_genotypes), Stress genotypes (stress_genotypes), and/or Virulence genotypes (virulence_genotypes).)
The table below shows the correspondences between the AMRFinderPlus methods used to identify genotypes and the simplified genotype categories displayed by the Isolates Browser web interface:
| AMRFinderPlus Method | Genotype Category in the Isolates Browser web display |
Notes |
| ALLELEP | COMPLETE | "Complete" genes are sequences that have BLAST alignments that cover ≥ 90% of the reference protein in the Pathogen Detection Reference Gene Catalog (sometimes referred to as the AMRFinderPlus database). Specifically:
|
| ALLELEX | ||
| BLASTP | ||
| BLASTX | ||
| EXACTP | ||
| EXACTX | ||
| HMM | HMM | These are proteins that were found by HMM only, more distant to reference proteins than our BLAST cutoffs. (The HMM was hit above the cutoff, but there was not a BLAST hit that met standards for BLAST or PARTIAL. This does not have a suffix of "P" or "X" because only protein sequences are searched by HMM.) |
| INTERNAL_STOP | MISTRANSLATION | Indicates a stop codon was found within the BLASTX alignment of the nucleotide sequence to the reference protein. In the future this may be extended to include frame shifts (which are currently not directly detected by AMRFinderPlus). |
| PARTIALP | PARTIAL | "Partial" genes are identified by BLAST to cover > 50% but < 90% of the length of the reference sequence, and the BLAST alignment does not end at a contig boundary. The aligned region has > 90% identity to the reference protein (default cutoff). For some genes, however, the sequence identity cutoff may be higher or lower, based on manual curation.. |
| PARTIALX | ||
| PARTIAL_CONTIG_ENDP | PARTIAL_END_OF_CONTIG | "Partial end of contig" genes are "partial" alignments that end at contig boundaries, indicating that they are more likely to have been split by a sequencing or assembly issue. Like "partial" genes, these are identified by BLAST to cover > 50% but < 90% of the length of the reference sequence. The aligned region has > 90% sequence identity to the reference (default cutoff). For some genes, however, the sequence identity cutoff may be higher or lower, based on manual curation. |
| PARTIAL_CONTIG_ENDX | ||
| POINTN | POINT | Point mutation identified by BLAST:
|
| POINTP | ||
| POINTX |
Data Retention and History Tracking
- Pathogen Detection Isolates Browser data retention
- Pathogen data and analysis results continue to evolve
- Three critical data objects are tracked
- Accession.Versions are used to track changes
- Two interactions that may not present the most up-to-date information
- Two states for data retention besides the most-up-to date version
- Example scenarios: responses to requests for previous versions of data that are older than 30 days
- Shared URLs are valid for 60 days
- Data retention policy for ftp
- MicroBIGG-E data retention
- Reference data retention (Reference Gene Catalog, Reference Gene Hierarchy, Reference HMM Catalog, and AMRFinderPlus database)
Pathogen Reference Data and Analysis Results Continue to Evolve
- Unlike many other databases and resources at NCBI, the Pathogen Detection Project was designed to provide updates of analyses in real-time. Therefore, the content of the resource may be updated multiple times per day. For any given pathogen isolate, organism group, or SNP cluster, the Pathogen Detection Browsers display, by default, the most current data and analysis results, including the relationships among isolates that have been calculated by the data processing pipeline. Therefore, for most uses of the browsers, the latest data are being presented. Of the browsers only the isolates browser retains some tracking of history as described below. The Reference Browsers (Reference Gene Catalog, Reference Gene Hierarchy, and Reference HMM Catalog) all only show data for the most recent release. A complete history is maintained on the FTP site. See AMRFinderPlus Reference Data Retention for details
Isolates Browser data retention
Three critical Isolates Browser data objects are tracked
- The system tracks versions for three critical data objects: The organism group is the entire package of new isolate updates, which could consist of both new or updated assemblies as well as new or updated clusters. Updates of each organism group could occur as frequently as every 24 hours, and as each organism group is independent of one another, multiple organism groups could be updated in a given day.
Accession.Versions are used to track changes
- The Pathogen Detection Project assigns an accession.version to each isolate genome assembly, organism group, and SNP cluster in order to track changes to the pathogens data and analysis results. For example:
- The Pathogen Detection Target ("PDT" accession.version) is the genome assembly for an individual isolate.
A new version of a PDT record indicates a change in the assembly. - The Pathogen Detection SNP cluster ("PDS" accession.version) is a group of isolates that are closely related, based on the SNP distance between their genome assemblies as calculated by the Pathogen Detection Project data processing pipeline.
A new version of a PDS record may indicate changes such as the following:
- The SNP cluster changed its membership.
- Some of its SNP distances have changed among the isolates that are members of the SNP cluster
- The Pathogen Detection Group ("PDG" accession.version) is also known as an organism group.
A new version of a PDG record includes additions or deletions of isolates, changes to isolate assemblies. All of these isolate assembly changes may or may not include changes to SNP clusters (additions, deletions, modifications). The Pathogens Detection Project retains the most recent 300 versions of a PDG.
- Technical note: An organism group (PDG) contains one or more targets (PDTs). A PDT is a member of zero or one SNP cluster (PDS), and never more than one cluster. A SNP cluster is composed of two or more PDTs, and each ach PDS is completely contained within a PDG.
- The Pathogen Detection Target ("PDT" accession.version) is the genome assembly for an individual isolate.
Two interactions that may not present the most up to date information
- As noted under Pathogen Data and Analysis Results Continue to Evolve, the latest data are presented by default for most uses of the browser. There are only two specific user interactions with the browser that may not present the most up-to-date information: 1) searches with specific accession.versions of one of the three objects mentioned above (PDT, PDG, PDS) that are from older analyses, and 2) the "share URL" button on the SNP Tree Viewer. For both of these cases there is a data retention policy is place that culls older data (i.e., removes versions of isolates, organism groups, and SNP clusters that were retired more than 30 days ago) so that the system does not need to retain every single piece of data ever calculated.
Two states for data retention besides the most up to date version
- There are two states for data retention besides the most up to date version. These include a window of 30 days where older data can be viewed, including the SNP tree as it looked for that particular version, and beyond that, the interface will present the user with links to the most current versions of that data.
Example scenarios: requests for previous versions of data that are older than 30 days
- If you try to view previous versions of the data, the following scenarios can occur:
- If you are requesting an accesion/version that is older than the 30 day retention period, you can no longer see the content (e.g., phylogenetic tree, SNP distances, metadata) for a target or cluster. However, the Pathogen Browser will indicate the current version of a requested target or cluster. It can also help you find successor target or cluster(s) if the requested target or cluster no longer exists. These hints are displayed at the top of the Browser.
- For example, if you enter PDT000000625.5 in the Search Isolates box, you get a message like this:
Record PDT000000625.5 replaced by PDG000000002.1212/PDT000000625.6. The system is directing you to the newer version PDT000000625.6 published in PDG version PDG000000002.1212.
- For example, if you enter PDT000000625.5 in the Search Isolates box, you get a message like this:
- If the requested cluster no longer exists, then a list of one or more successor clusters may be presented. The Pathogen Browser determines the past target membership of the requested cluster and traces forward to the current clusters that contain those targets. This allows forward tracking of a cluster when the cluster has split or merged over time, or has been completely replaced.
- For example, if you enter PDS000029842.1 in the Search Isolates box, you get message like this:
SNP cluster(s) succeeded by PDG000000002.1212/PDS000032550.9.
- For example, if you enter PDS000029842.1 in the Search Isolates box, you get message like this:
- Occasionally a target may be withdrawn (taken out of service) usually as a result of data retraction by a submitter. When you request such a target, the Pathogen Browser will try to direct you to the cluster (or its successor(s)) that once included the target as a member.
- For example, if you enter PDT000111278.1 you will get a message like this:
Record removed: PDT000111278.1 SNP cluster(s) succeeded by PDG000000002.1212/PDS000028815.20.
- For example, if you enter PDT000111278.1 you will get a message like this:
- Using a shared URL that you either made in the past or got from a collaborator could result in any one of the following, depending on the age of the shared URL and whether the URL refers to actual content within the 30d retention period:
- A tree viewer display (if the URL refers to current data, or to data that is still available as a result of the 30 day retention policy)
OR - A history tracking message such as the ones in the examples above (if the URL is less than 60 days old and refers to data that is no longer available in its previous form)
OR - A message saying the URL has expired (if the URL is more than 60 days old). In that case, if you are still interested in viewing the isolate, organism group, or SNP cluster that is cited in the URL, you can enter the corresponding PDT*, PDG*, or PDS* accession number in the Isolates Browser to access the most recent version of the data.
- A tree viewer display (if the URL refers to current data, or to data that is still available as a result of the 30 day retention policy)
- If you are requesting an accesion/version that is older than the 30 day retention period, you can no longer see the content (e.g., phylogenetic tree, SNP distances, metadata) for a target or cluster. However, the Pathogen Browser will indicate the current version of a requested target or cluster. It can also help you find successor target or cluster(s) if the requested target or cluster no longer exists. These hints are displayed at the top of the Browser.
Shared URLs are valid for 60 days
- A "Share" button is available in the SNP Tree Viewer display (as shown in part C of the illustrated example of a SNP Tree Viewer display). It produces a URL that captures your customized view of the tree, which can then be copied and shared with others to reproduce the same view.
- The URL is temporary, remaining valid for 60 days:
- For the first 30 days, the URL will open the customized display, showing the isolates you selected and any other customizations you made to the view.
- For the second 30 days, the URL continues to be valid, but during that time, it will only show a link to the default display for the most recent version of the SNP cluster. That is, the URL will not open the original customized view, but instead will redirect to a version of the phylogenetic distance tree that reflects the most recent for the tree.
Isolates browser data published to FTP are also subject to retention policies
- Progressive retention policy:
- Every publication within 30 days
- One publication per week after 30 days but within 6 months
- One publication per month after 6 months but within 1 year
- One publication per year thereafter
MicroBIGG-E data retention
- MicroBIGG-E and the
ncbi-pathogen-detect.pdbrowser.microbiggeBigQuery table only show the most recent available data. NCBI Pathogen Detection does not archive past results.
Reference browser data retention
- The three reference browsers (Reference Gene Catalog, Reference Gene Hierarchy, and Reference HMM Catalog) only show data from the most recent release. The release version is shown above the upper left of the table (e.g., db version: 2022-08-09.1).
- All release versions and the data behind them are archived on the Pathogen Detection FTP site at https://ftp.ncbi.nlm.nih.gov/pathogen/Antimicrobial_resistance/AMRFinderPlus/database. This includes all the data files for the Reference Gene Catalog, the Reference Gene Hierarchy, and the Reference HMM Catalog, the AMRFinderPlus database, a change log (changes.txt), and the data behind the browsers in tab-delimited text format. See the AMRFinderPlus wiki for a detailed list of files and the formats of those files.
Log of Changes to Pathogen Detection Project
AMRFinderPlus database change log
- JANUARY 2024 - Organism Group change: The Enterococcus faecium organism group (PDG000000071) has been split into its constituent species-specific organism groups as follows: Enterococcus faecium remains PDG000000071, and Enterococcus hirae has been split to a new organism group (PDG000000122). In addition, the wgMLST locus found QC threshold has been lowered for Enterococcus faecium thus admitting more isolates into the SNP clustering process.
- OCTOBER 2023 - Released the MicroBIGG-E Map (MicroBIGG-E Map)
- MARCH 2023 - Organism Group change:
The Enterobacter organism group (PDG000000028) has been split into its constituent species-specific organism groups:
Enterobacter has been renamed to Enterobacter cloacae (remains PDG000000028)
New organism groups have been created by splitting off the respective isolates as follows:
-
Enterobacter asburiae (PDG000000106)
Enterobacter bugandensis (PDG000000111)
Enterobacter cancerogenus (PDG000000107)
Enterobacter chengduensis (PDG000000118)
Enterobacter chuandaensis (PDG000000119)
Enterobacter hormaechei (PDG000000105)
Enterobacter kobei (PDG000000108)
Enterobacter ludwigii (PDG000000109)
Enterobacter mori (PDG000000110)
Enterobacter roggenkampii (PDG000000116)
Enterobacter sichuanensis (PDG000000117)
Enterobacter soli (PDG000000112)
Kosakonia oryzendophytica (PDG000000113)
Kosakonia oryziphila (PDG000000114)
Phytobacter massiliensis (PDG000000115)
- NOVEMBER 2022 - Organism Group change:
The Neisseria organism group (PDG000000032) has been split into its constituent species-specific organism groups:
Neisseria has been renamed to Neisseria gonorrhoeae (remains PDG000000032)
New organism groups have been created by splitting off the respective isolates as follows:
-
Neisseria meningitidis (PDG000000097)
Neisseria lactamica (PDG000000095)
Neisseria polysaccharea (PDG000000096)
- AUGUST 2022 - Isolates Browser and MicroBIGG-E data available in Google Cloud Platform.
- JUNE 2022 - Organism Group change: The "Listeria" organism group has been reorganized. All Listeria monocytogenes isolates continue to be clustered as before under "Listeria" (PDG000000001). Listeria innocua isolates can now be found in a new organism group called "Listeria innocua" (PDG000000091). More organism groups are currently undergoing evaluation for splitting.
- OCTOBER 2021 - Released the Pathogen Detection Reference Gene Hierarchy (Reference Gene Hierarchy).
- AUGUST 2021 - Changed the Isolation Type / epi_type attribute that affects min-same/min-diff computation to default to NULL instead of environmental/other in the Isolates Browser, MicroBIGG-E, and SNP Tree Viewer. This means that the min-same/min-diff values for an isolate may show n/a, and other min-same/min-diff values for isolates may change. Added the Computed types / computed_types field to the Isolates Browser and SNP Tree Viewer.
- MARCH 2021 - Released the Pathogen Detection Reference HMM Catalog (Reference HMM Catalog).
- JULY 2020 - Released the Microbial Browser for Genetic and Genomic Identification (MicroBIGG-E).
- FEBRUARY 2020 - Added five new data fields to the Isolates Browser, including: amrfinderplus_analysis_type, amrfinderplus_version, refgene_db_version, stress_genotypes, and virulence_genotypes. Added a "Share" function to the Isolates Browser; it produces a URL that captures your search strategy, which can then be copied and shared with others to execute the search. (The results of the search will change over time as new data become available.) In the SNP Tree Viewer, enhanced the Search & Highlight in Tree function so it now searches all labels that are currently displayed by the SNP Tree Viewer, including custom labels you might have added to the tree. (Previously, the "Search in tree" function searched only the default set of labels.)
- FALL 2019 - Released AMRFinderPlus with blacklist options for certain genes ubiquitous in some species. Incorporated first fungal pathogen: Candida auris.
- SUMMER 2019 - Released AMRFinderPlus with additional virulence and stress response genes. Publication by Feldgarden et al. (2019), describing the NCBI AMRFinder tool, became available in Antimicrob Agents Chemother as an e-pub ahead of print (PubMed PMID: 31427293; Full text at AAC, doi: 10.1128/AAC.00483-19).
- SPRING 2019 - Released Antimicrobial Resistance (AMR) resources, including an AMR landing page, AMR resources page, and the Pathogen Detection Reference Gene Catalog. Preprint by Feldgarden et al. (2019), describing the NCBI AMRFinder tool, became available in BioRxiv, doi.org/10.1101/550707. Released a new service that enables you to receive Automatic e-mail notifications of new data. Released the Pathogens help document.
- SUMMER 2018 - Switch to wgMLST clustering begins.
- MAY 2018 - Updated version of pathogen browser to improve navigation within larger SNP trees.
- FEB 2018 - Beta release of Pathogen Browser.
- Jul 2017 - Alpha release of new Pathogen Browser.
- MAY 2017 - Development of rapid reports based on SKESA/wgMLST.
- DEC 2016 - Addition of antimicrobial resistant genotypes/phenotypes to Pathogen Browser.
- MAY 2016 - NCBI Pathogen Browser released.
- DEC 2015 - Automated delivery of SNP trees to FTP from NCBI pipeline.
- SPRING 2015 - Whitehouse initiative on Combatting Antibiotic Resistant Bacteria (CARB), includes building a national database of resistant pathogens; NCBI working on a system to identify resistance genes per isolate and the system to capture resistance phenotypes per isolate.
- FALL 2014 - Automated delivery of NCBI analysis results, k-mer trees, work begins on detection of AMR genes/proteins.
- SUMMER 2013 - Data starts to arrive at NCBI in summer of 2013.
- JAN 2013 - NCBI begins modeling data systems/pipelines for Pathogen Detection.
- MAR 2012 - NCBI begins integration of analysis pipeline into gpipe, k-mer analysis, assembly, SNP calling.
- OCT 2011 - NCBI begins R&D into a framework for analysis following the 2nd Global Microbial Identifier (GMI) meeting.
- SEP 2011 - 1st Global Microbial Identifier (GMI) meeting: http://www.globalmicrobialidentifier.org/, http://science.sciencemag.org/content/333/6051/1818.full.
References
NCBI Publications/Methods used by the Pathogen Detection Project
Third party Publications/Methods used by the Pathogen Detection Project
Publications from Other Sources using the Pathogen Detection Browser
Publications from External Labs using the Pathogen Detection Browser
Presentations about the Pathogen Detection Project
References about the Genomics for Food Safety (GenFS) initiative
References about the FDA GenomeTrakr project and WGS activities
References about the CDC PulseNet network and WGS activities
References about Public Health England WGS activities
Other related references
References on antimicrobial resistance, including AMRFinder
Citing the NCBI Pathogen Detection Project
- The NCBI Pathogen Detection Project [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. 2016 May [cited YYYY MMM DD]. Available from: https://www.ncbi.nlm.nih.gov/pathogens/
NCBI Publications/Methods used by the Pathogen Detection Project
- Souvorov A and Agarwala R. SAUTE: sequence assembly using target enrichment. BMC Bioinformatics. 2021 Jul 21;22(1):375. doi: 10.1186/s12859-021-04174-9. PubMed PMID: 34289805; Full text at BMC.
- Souvorov A, Agarwala R, Lipman DJ. SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018 Oct 4;19(1):153. doi: 10.1186/s13059-018-1540-z. PubMed PMID: 30286803; Full text in PubMed Central PMCID: PMC6172800; Full text at BMC.
- Cherry JL. A practical exact maximum compatibility algorithm for reconstruction of recent evolutionary history. BMC Bioinformatics 2017 Feb 23;18(1):127. doi: 10.1186/s12859-017-1520-4. PubMed PMID: 28231758; Full text in PubMed Central PMCID: PMC5324209; Full text at BMC.
The AMRFinderPlus software is used to identify antimicrobial resistance genes plus select virulence, biocide, metal, and stress resistance genes. Software is available at https://github.com/ncbi/amr/wiki
- Feldgarden M, Brover V, Fedorov B, Haft DH, Prasad AB, Klimke W. Curation of the AMRFinderPlus databases: applications, functionality and impact. Microb Genome. 2022 Jun;8(6). doi: 10.1099/mgen.0.000832. PubMed PMID: 35675101; Full text at Microbial Genomics.
- Feldgarden M, Brover V, Gonzalez-Escalona N, Frye JG, Haendiges J, Haft DH, Hoffmann M, Pettengill JB, Prasad AB, Tillman GE, Tyson GH, Klimke W. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci Rep. 2021 June 16;11(1):12728. https://doi.org/10.1038/s41598-021-91456-0. PubMed PMID: 34135355; Full text at Nature Scientific Reports.
- Feldgarden M, Brover V, Haft DH, Prasad AB, Slotta DJ, Tolstoy I, Tyson GH, Zhao S, Hsu C-H, McDermott PF, Tadesse DA, Morales C, Simmons M, Tillman G, Wasilenko J, Folster JP, Klimke W. Validating the NCBI AMRFinder Tool and Resistance Gene Database Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of NARMS Isolates. Antimicrobial Agents and Chemotherapy. 2019 Nov 1;63(11). doi: e10.1128/AAC.00483-19 PubMed PMID: 31427293; Full text in PubMed Central PMCID: PMC6811410; Full text at AAC.
- Haft DH, DiCuccio M, Badretdin A, Brover V, Chetvernin V, O'Neill K, Li W, Chitsaz F, Derbyshire MK, Gonzales NR, Gwadz M, Lu F, Marchler GH, Song JS, Thanki N, Yamashita RA, Zheng C, Thibaud-Nissen F, Geer LY, Marchler-Bauer A, Pruitt KD. RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Research. 2018 Jan 4;46(D1):D851-D860. doi: 10.1093/nar/gkx1068 PubMed PMID: 29112715; Full text in PubMed Central PMCID: PMC5753331; Full text at Oxford University Press.
Third Party Publications/Methods used by the Pathogen Detection Project
- Zhang S, den Bakker HC, Li S, Chen J, Dinsmore BA, Lane C, Lauer AC, Fields PI, Deng X. SeqSero2: Rapid and Improved Salmonella Serotype Determination Using Whole-Genome Sequencing Data. Appl Environ Microbiol. 2019 Nov 14;85(23):e01746-19. doi: 10.1128/AEM.01746-19. PubMed PMID: 31540993; Full text at PubMed Central PMCID: PMC6856333.
- Zhang S, Yin Y, Jones MB, Zhang Z, Deatherage Kaiser BL, Dinsmore BA, Fitzgerald C, Fields PI, Deng X. Salmonella serotype determination utilizing high-throughput genome sequencing data. J Clin Microbiol. 2015 May;53(5):1685-92. doi: 10.1128/JCM.00323-15. Epub 2015 Mar 11. PubMed PMID: 25762776; Full text at PubMed Central PMCID: PMC4400759.
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019 Aug 2;37:907-915. doi:10.1038/s41587-019-0201-4; PubMed PMID:31375807; Full text at PubMed Central PMCID: PMC7605509.
Publications from other sources using the Pathogen Detection Browser
- ProPublica (2021) Uses NCBI Pathogen Detection to look at Salmonella Infantis problem in food products. https://www.propublica.org/article/how-propublica-used-genomic-sequencing-data-to-track-an-ongoing-salmonella-outbreak https://www.propublica.org/article/salmonella-chicken-usda-food-safety
Publications from External Labs using the Pathogen Detection Browser
- Schwan CL, Dallman TJ, Cook PW, Vipham J (2022) A case report of Salmonella enterica serovar Corvallis from environmental isolates from Cambodia and clinical isolates in the UK. Access Microbiology: Vol4(1) https://doi.org/10.1099/acmi.0.000315
- Brown B, Allard M, Bazaco MC, Blankenship J, Minor T (2021) An economic evaluation of the Whole Genome Sequencing source tracking program in the U.S. PLoS ONE 16(10): e0258262. https://doi.org/10.1371/journal.pone.0258262
- Ladd-Wilson SG, Morey K, Turpen L, DeMarco K,Van Der Veen G,Fontana JL, Dannenhoffer RL, Tenney K, Kutumbaka KK, Samadpour M, Cieslak PR. Escherichia coli O157:H7 Cluster Associated With Deer Harvested at a Single Wildlife Hunting Area, Oregon, 2017. Full text at Public Health Reports.
- Worley JN, Javkar K, Hoffmann M, Hysell K, Garcia-Williams A, Tagg K, Kanjilal S, Strain E, Pop M, Allard M, Francois Watkins L, Bry L. Genomic Drivers of Multidrug-Resistant Shigella Affecting Vulnerable Patient Populations in the United States and Abroad. PubMed PMID: 33500335; Full text at mBio.
- Carey J, Cole J, Venkata SLG, Hoyt H, Mingle L, Nicholas D, Musser KA, Wolfgang WJ. Genomic Epidemiology of Historical Clostridium perfringens Outbreaks in New York State Using Two Web-based Platforms: National Center for Biotechnology Information-Pathogen Detection and FDA-GalaxyTrakr. PubMed PMID: 33177125; Full text at Journal of Clinical Microbiology.
- Pettengill J, Markell A, Conrad A, Carleton H, Beal J, Rand H, Musser S, Brown E, Allard M, Huffman J, Harris S, Wise M, Locas A. A multinational listeriosis outbreak and the importance of sharing genomic data. Full text at The Lancet.
- Worley J, Delaney ML, Cummins CK, DuBois A, Klompas, Bry L. Genomic determination of relative risks for Clostridioides difficile infection from asymptomatic carriage in ICU patients. PubMed PMID: 32676661; Full text at Clin Infect Dis.
- Ladd-Wilson SG, Morey K, Koske SE, Burkhalter B, Bottichio L, Brandenburg J,Fontana J, Tenney K, Kutumbaka KK, Samadpour M, Kreil K, Cieslak PR. Notes from the Field: Multistate Outbreak of Salmonella Agbeni Associated with Consumption of Raw Cake Mix - Five States, 2018. PubMed PMID: 31465317; Full text in PubMed Central PMCID: PMC6715262; Full text at MMWR.
- Ezernitchi AV, Sirotkin E, Danino D, Agmon V, Valinsky L,Rokney A. Azithromycin non-susceptible Shigella circulating in Israel, 2014-2016. PLoS One. 2019 Oct 18;14(10):e0221458. doi: 10.1371/journal.pone.0221458. eCollection 2019. PubMed PMID: 31465317; Full text in PubMed Central PMCID: PMC6799884; Full text at PLoS ONE.
- Sekyere JO, and Reta MA. Genomic and Resistance Epidemiology of Gram-Negative Bacteria in Africa: a Systematic Review and Phylogenomic Analyses from a One Health Perspective. PubMed PMID: 33234606; Full text at mSystems.
- Diemert S, Yan T. Clinically Unreported Salmonellosis Outbreak Detected via Comparative Genomic Analysis of Municipal Wastewater Salmonella Isolates. Appl Environ Microbiol. 2019 May 2;85(10). pii: e00139-19. doi: 10.1128/AEM.00139-19. Print 2019 May 15 PubMed PMID: 30902850; Full text in PubMed Central PMCID: PMC6498150; Full text at Applies and Environmental Microbiology.
Presentations about the Pathogen Detection Project
- NCBI Webinar: "Introducing the NCBI Pathogen Detection Isolates Browser," March 21, 2018 (webinar announcement/description; recording of webinar (31:23 minutes); slides and questions/answers)
- American Society for Microbiology Microbe Conference in San Francisco, CA, June 20-24, 2019 .
The following presentations by the NCBI Pathogen Detection Project Group and the NCBI Taxonomy Group are available on the NCBI FTP site: https://ftp.ncbi.nlm.nih.gov/pub/factsheets/ASM2019_Talks_Posters/:
- ASM Microbe Conference in Washington, DC, June 20-24, 2022
- American Society for Microbiology NGS 2022 in Baltimore, MD, Oct 16-19, 2022
- PD team participated in a workshop at ASM NGS 2022 which included some projects that demonstrate how to use our resources in the cloud.
References about the Genomics for Food Safety (GenFS) initiative
- Stevens EL, Carleton HA, Beal J, Tillman GE, Lindsey RL, Lauer AC, Pightling A, Jarvis KG, Ottesen A, Ramachandran P, Hintz L, Katz LS, Folster JP, Whichard JM, Trees E, Timme RE, McDERMOTT P, Wolpert B, Bazaco M, Zhao S, Lindley S, Bruce BB, Griffin PM, Brown E, Allard M, Tallent S, Irvin K, Hoffmann M, Wise M, Tauxe R, Gerner-Smidt P, Simmons M, Kissler B, Defibaugh-Chavez S, Klimke W, Agarwala R, Lindsay J, Cook K, Austerman SR, Goldman D, McGARRY S, Hale KR, Dessai U, Musser SM, Braden C. Use of Whole Genome Sequencing by the Federal Interagency Collaboration for Genomics for Food and Feed Safety in the United States. J Food Prot. 2022. May 1;85(5):755-772. doi: 10.4315/JFP-21-437. PubMed PMID: 35259246;
- Timme RE, Rand H, Shumway M, Trees EK, Simmons M, Agarwala R, Davis S, Tillman GE, Defibaugh-Chavez S, Carleton HA, Klimke WA, Katz LS. Benchmark datasets for phylogenomic pipeline validation, applications for foodborne pathogen surveillance. PeerJ 2017 Oct 6;5:e3893. doi: 10.7717/peerj.3893. eCollection 2017. PubMed PMID: 29372115; Full text in PubMed Central PMCID: PMC5782805; Full text at PeerJ.
References about the FDA GenomeTrakr project and WGS activities
- New Era of Smarter Food Safety TechTalk Podcast. TechTalk Podcast Episode 2: Whole Genome Sequencing in the New Era of Smarter Food Safety. MP3 link.
- FDA Publishes Report on Focus Areas of Regulatory Science. FDA Publishes Report on Focus Areas of Regulatory Science. PDF Version.
- Timme RE, Rand H, Sanchez Leon M, Hoffmann M, Strain E, Allard M, Roberson D, Baugher JD. GenomeTrakr proficiency testing for foodborne pathogen surveillance: an exercise from 2015. Microb Genom. 2018 Jul;4(7). doi: 10.1099/mgen.0.000185. Epub 2018 Jun 15. PubMed PMID: 29906258; Full text in PubMed Central PMCID: PMC6113870; Full text at Microbiology Society
- Timme RE, Rand H, Sanchez Leon M, Hoffmann M, Strain E, Allard M, Roberson D, Baugher JD. GenomeTrakr proficiency testing for foodborne pathogen surveillance: an exercise from 2015. Microb Genom. 2018 Jul;4(7). doi: 10.1099/mgen.0.000185. Epub 2018 Jun 15. PubMed PMID: 29906258; Full text in PubMed Central PMCID: PMC6113870; Full text at Microbiology Society
- Allard MW, Strain E, Melka D, Bunning K, Musser SM, Brown EW, Timme R. Practical Value of Food Pathogen Traceability through Building a Whole-Genome Sequencing Network and Database. J Clin Microbiol. 2016 Aug;54(8):1975-83. doi: 10.1128/JCM.00081-16. Epub 2016 Mar 23. Review. PubMed PMID: 27008877; Full text in PubMed Central PMCID: PMC4963501; Full text at American Society for Microbiology
- Allard MW, Strain E, Rand H, Melka D, Correll WA, Hintz L, Stevens E, Timme R, Lomonaco S, Chen Y, Musser SM, Brown EW. Whole genome sequencing uses for foodborne contamination and compliance: Discovery of an emerging contamination event in an ice cream facility using whole genome sequencing. Infect Genet Evol. 2019 Sep;73:214-220. doi: 10.1016/j.meegid.2019.04.026. Epub 2019 Apr 27. PubMed PMID: 31039448; Full text at Infection, Genetics and Evolution
- Trinetta V, Magossi G, Allard MW, Tallent SM, Brown EW, Lomonaco S. Characterization of Salmonella enterica Isolates From Selected U.S. Swine Feed Mills by Whole-Genome Sequencing. Foodborne Pathog Dis. 2020 Feb;17(2):126-136. doi: 10.1089/fpd.2019.2701. Epub 2019 Nov 8. PubMed PMID: 31702400; Full text at Foodborne Pathog Dis.
- Timme RE, Wolfgang WJ, Balkey M, Venkata SLG, Randolph R, Allard M, Strain E. Optimizing open data to support one health: best practices to ensure interoperability of genomic data from bacterial pathogens. One Health Outlook. 2020 Epub 2020 Oct 19. doi: 10.1186/s42522-020-00026-3. PubMed PMID: 33103064; Full text at One Health Outlook.
References about the CDC PulseNet network and WGS activities
- Armstrong GL, MacCannell DR, Taylor J, Carleton HA, Neuhaus EB, Bradbury RS, Posey JE, Gwinn M. Pathogen Genomics in Public Health. N Engl J Med. 2019 Dec 26;381(26):2569-2580. doi: 10.1056/NEJMsr1813907.PubMed PMID: 31881145; Full text at New England Journal of Medicine
- Nadon C, Van Walle I, Gerner-Smidt P, Campos J, Chinen I, Concepcion-Acevedo J, Gilpin B, Smith AM, Man Kam K, Perez E, Trees E, Kubota K, Takkinen J, Nielsen EM, Carleton H; FWD-NEXT Expert Panel. PulseNet International: Vision for the implementation of whole genome sequencing (WGS) for global food-borne disease surveillance. Euro Surveill. 2017 Jun 8;22(23). pii: 30544. doi: 10.2807/1560-7917.ES.2017.22.23.30544. Review. PubMed PMID: 28662764; Full text in PubMed Central PMCID: PMC5479977; Full text at Eurosurveillance
- Announcement: 20th Anniversary of PulseNet: the National Molecular Subtyping Network for Foodborne Disease Surveillance - United States, 2016. MMWR Morb Mortal Wkly Rep. 2016 Jun 24;65(24):636. doi: 10.15585/mmwr.mm6524a5.. PubMed PMID: 27337605; Full text at CDC
- Jackson BR, Tarr C, Strain E, Jackson KA, Conrad A, Carleton H, Katz LS, Stroika S, Gould LH, Mody RK, Silk BJ, Beal J, Chen Y, Timme R, Doyle M, Fields A, Wise M, Tillman G, Defibaugh-Chavez S, Kucerova Z, Sabol A, Roache K, Trees E, Simmons M, Wasilenko J, Kubota K, Pouseele H, Klimke W, Besser J, Brown E, Allard M, Gerner-Smidt P. Implementation of Nationwide Real-time Whole-genome Sequencing to Enhance Listeriosis Outbreak Detection and Investigation. Clin Infect Dis. 2016 Aug 1;63(3):380-6. doi: 10.1093/cid/ciw242. Epub 2016 Apr 18. PubMed PMID: 27090985; Full text in PubMed Central PMCID: PMC4946012; Full text at Oxford Academic.
References about Public Health England WGS activities
- Chattaway MA, Dallman TJ, Larkin L, Nair S, McCormick J, Mikhail A, Hartman H, Godbole G, Powell D1, Day M, Smith R, Grant K. The Transformation of Reference Microbiology Methods and Surveillance for Salmonella With the Use of Whole Genome Sequencing in England and Wales. Front Public Health. 2019 Nov 21;7:317. doi: 10.3389/fpubh.2019.00317. eCollection 2019. PubMed PMID: 31824904; Full text in PubMed Central PMCID: PMC6881236; Full text at Frontiers in Public Health.
Other related references
- Global Microbial Identifier (GMI) initiative:
Kupferschmidt K. Epidemiology. Outbreak detectives embrace the genome era. Science. 2011 Sep 30;333(6051):1818-9. doi: 10.1126/science.333.6051.1818. PubMed PMID: 21960605; Full text at Science Magazine
GMI website: http://www.globalmicrobialidentifier.org/Note: The Global Microbial Identifier (GMI) initiative is a grassroots attempt to build a global system of DNA genome databases for microbial and infectious disease identification and diagnostics. Sequencing projects that are flagged with the 'GMI' keyword can be found in the NCBI BioProject database via this search: https://www.ncbi.nlm.nih.gov/bioproject/?term=GMI[keyword] - FDA-ARGOS:
Sichtig H, Minogue T, Yan Y, Stefan C, Hall A, Tallon L, Sadzewicz L, Nadendla S, Klimke W, Hatcher E, Shumway M, Aldea DL, Allen J, Koehler J, Slezak T, Lovell S, Schoepp R, Scherf U. FDA-ARGOS is a database with public quality-controlled reference genomes for diagnostic use and regulatory science. Nat Commun.. 2019 Jul 25;10(1):3313. doi: 10.1038/s41467-019-11306-6. PubMed PMID: 31346170; Full text in PubMed Central PMCID: PMC6658474; Full text at Nature Publishing Group
References on antimicrobial resistance
- Feldgarden M, Brover V, Fedorov B, Haft DH, Prasad AB, Klimke W. Curation of the AMRFinderPlus databases: applications, functionality and impact. Microb Genome. 2022 Jun;8(6). doi: 10.1099/mgen.0.000832. PubMed PMID: 35675101; Full text at Microbial Genomics.
- Feldgarden M, Brover V, Gonzalez-Escalona N, Frye JG, Haendiges J, Haft DH, Hoffmann M, Pettengill JB, Prasad AB, Tillman GE, Tyson GH, Klimke W. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci Rep. 2021 June 16;11(1):12728. https://doi.org/10.1038/s41598-021-91456-0. PubMed PMID: 34135355; Full text at Nature Scientific Reports.
- Feldgarden M, Brover V, Haft DH, Prasad AB, Slotta DJ, Tolstoy I, Tyson GH, Zhao S, Hsu CH, McDermott PF, Tadesse DA, Morales C, Simmons M, Tillman G, Wasilenko J, Folster JP, Klimke W. Validating the NCBI AMRFinder Tool and Resistance Gene Database Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of NARMS Isolates. Antimicrob Agents Chemother 2019 Aug 19. pii: AAC.00483-19. doi: 10.1128/AAC.00483-19. [Epub ahead of print] PubMed PMID: 31427293; Full text at AAC.
- Papan C., et al. Combined antibiotic stewardship and infection control measures to contain the spread of linezolid-resistant Staphylococcus epidermidis in an intensive care unit.Antimicrob Resist Infect Control. 2021 June https://doi.org/10.1186/s13756-021-00970-3. Full text at Antimicrob Resist Infect Control. PubMed PMID: 34193293 ;
- Wee S.K., et al. Draft Genome Sequence of Enterobacter hormaechei subsp. steigerwaltii Strain BEI01.Microbiol Resour Announc. 2021 July https://doi.org/10.1128/mra.00406-21. Full text at Microbiol Resour Announc. PubMed PMID: 34264109 ;
- Zhang A-N, et al. An omics-based framework for assessing the health risk of antimicrobial resistance genes.Nature Communications 2021 August https://doi.org/10.1038/s41467-021-25096-3. Full text at Nature Communications.PubMed PMID: 34362925 ;
- David J, et al. The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities.NAR 2020 January https://doi.org/10.1093/nar/gkz943. Full text at Nucleic Acids Research.PubMed PMID: 31667520 ;
- Cole SD, Peak L, Tyson GH, Reimschuessel R, Ceric O, Rankin SC. New Delhi Metallo-beta-Lactamase-5-producing Escherichia coli in Companion Animals, United States. Emerg Infect Dis. 2020 Feb https://doi.org/10.3201/eid2602.191221. Full text at Emerging Infectious Diseases.
- Tyson GH, Li C, Hsu CH, Bodeis-Jones S, McDermott PF. Diverse Fluoroquinolone Resistance Plasmids From Retail Meat E. coli in the United States. Front Microbiol. 2019 Dec 5;10:2826. doi: 10.3389/fmicb.2019.02826. eCollection 2019. PubMed PMID: 31866986 ; Full text in PubMed Central PMCID: PMC6906146 ; Full text at Frontiers in Microbiology.
- Bush K. Past and Present Perspectives on β-Lactamases. Antimicrob Agents Chemother 2018 Sep 24;62(10). pii: e01076-18. doi: 10.1128/AAC.01076-18. Print 2018 Oct. Review. PubMed PMID: 30061284; Full text in PubMed Central PMCID: PMC6153792.
- Mack AR, Barnes MD, Taracila MA, Hujer AM, Hujer KM, Cabot G, Feldgarden M, Haft DH, Klimke W, van den Akker F, Vila AJ, Smania A, Haider S, Papp-Wallace KM, Bradford PA, Rossolini GM, Docquier JD, Frère JM, Galleni M, Hanson ND, Oliver A, Plésiat P, Poirel L, Nordmann P, Palzkill TG, Jacoby GA, Bush K, Bonomo RA. A standard numbering scheme for class C β-Lactamases. Antimicrob Agents Chemother 2019 Nov 11. pii: AAC.01841-19. doi: 10.1128/AAC.01841-19. [Epub ahead of print]. PubMed PMID: 31712217; Full text in Antimicrobial Agents and Chemotherapy.
- Partridge SR, Di Pilato V, Doi Y, Feldgarden M, Haft DH, Klimke W, Kumar-Singh S, Liu JH, Malhotra-Kumar S, Prasad A, Rossolini GM, Schwarz S, Shen J, Walsh T, Wang Y, Xavier BB. Proposal for assignment of allele numbers for mobile colistin resistance (mcr) genes. J Antimicrob Chemother 2018 2018 Oct 1;73(10):2625-2630. doi: 10.1093/jac/dky262. PubMed PMID: 30053115; Full text in PubMed Central PMCID: PMC6148208.
- Vasquez AM, Montero N, Laughlin M, Dancy E, Melmed R, Sosa L, Watkins LF, Folster JP, Strockbine N, Moulton-Meissner H, Ansari U, Cartter ML, Walters MS. Investigation of Escherichia coli Harboring the mcr-1 Resistance Gene - Connecticut, 2016. MMWR Morb Mortal Wkly Rep. 2016 Sep 16;65(36):979-80. doi: 10.15585/mmwr.mm6536e3. PubMed PMID: 27631346; Full text at CDC.
- Rehman MA, Yin X, Persaud-Lachhman MG, Diarra MS. First Detection of a Fosfomycin Resistance Gene, fosA7, in Salmonella enterica Serovar Heidelberg Isolated from Broiler Chickens. Antimicrob Agents Chemother. 2017 Jul 25;61(8). pii: e00410-17. doi: 10.1128/AAC.00410-17. Print 2017 Aug. PubMed PMID: 28533247; Full text in PubMed Central PMCID: PMC5527569; Full text at American Society for Microbiology.
- Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN, Doshi S, Courtot M, Lo R, Williams LE, Frye JG, Elsayegh T, Sardar D, Westman EL, Pawlowski AC, Johnson TA, Brinkman FS, Wright GD, McArthur AG. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017 Jan 4;45(D1):D566-D573. doi: 10.1093/nar/gkw1004. Epub 2016 Oct 26. PubMed PMID: 27789705; Full text in PubMed Central PMCID: PMC5210516; Full text at Oxford Academic.
- Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012 Nov;67(11):2640-4. doi: 10.1093/jac/dks261. Epub 2012 Jul 10. PubMed PMID: 22782487; Full text in PubMed Central PMCID: PMC3468078; Full text at Oxford Academic.
- Zankari E, Allesøe R, Joensen KG, Cavaco LM, Lund O, Aarestrup FM. PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J Antimicrob Chemother. 2017 Oct 1;72(10):2764-2768. doi: 10.1093/jac/dkx217. PubMed PMID: 29091202; Full text in PubMed Central PMCID: PMC5890747; Full text at Oxford Academic.
Contact information
If you would like to contact the NCBI Pathogen Detection team, please send an email to: pd-help@ncbi.nlm.nih.gov
Revised 18 Dec 2023
![]() Pathogen Detection Project help: pd-help@ncbi.nlm.nih.gov
Pathogen Detection Project help: pd-help@ncbi.nlm.nih.gov