

NCBI RefSeq Select
Introduction
The RefSeq Select dataset consists of a representative or “Select” transcript for every protein-coding gene. The transcript is chosen by an automated pipeline based on multiple selection criteria, which include prior use in clinical databases (e.g., Locus Reference Genomic), transcript expression, conservation of the coding region, transcript and protein length and concordance with the Swiss-Prot canonical isoform. The RefSeq Select transcript is usually well-supported by archived data, well-expressed, conserved and represents the biology of the gene.
Rationale
Many genes are represented by multiple RefSeq transcripts/proteins due to alternative splicing. These different transcripts may have distinct biological properties, such as differential expression or production of functionally distinct protein isoforms, whereas others may represent relatively rare forms that may or may not be functionally relevant. This complexity is problematic for analyses such as comparative genomics that often require just a single protein per gene, or exchange of clinical variant data where a focused dataset is preferred. RefSeq Select identifies suitable RefSeq transcripts and proteins for those purposes.
Taxonomic scope
Currently, RefSeq Select is available for the RefSeq human and mouse annotations, and for prokaryotic genomes. In the future, we plan to provide the RefSeq Select set for additional eukaryotes, including key model organisms and taxa with official nomenclature.
RefSeq Select in Prokaryotes
In August 2020, the scope of the RefSeq Select set was extended to include prokaryotic genomes. For prokaryotes, RefSeq Select is defined as proteins annotated on RefSeq reference and representative genomes. In most cases there is only one reference or representative genome per prokaryote species. RefSeq Select for prokaryotes is a useful way to explore inter-species protein diversity, especially for core proteins, but be aware that some strain-specific accessory proteins may not be included for some species. The RefSeq Select dataset is refreshed daily as the selection of prokaryote representative genomes is refined and individual genomes are re-annotated. It currently includes about one-third of the prokaryote RefSeq protein dataset. At present, Select data can be accessed via the Entez filter ("Bacteria"[Organism] OR "Archaea"[Organism] OR prokaryote[All Fields]) AND refseq_select[filter] in NCBI’s Protein databasee. Additional ways to access the data, including Blast databases, will be available soon.
RefSeq Select in Eukaryotes
Currently, all protein-coding genes in the human and mouse RefSeq annotations are in scope for the RefSeq Select set. Only the known (curated, with NM_ prefix) transcripts are designated as RefSeq Select. Note: The human RefSeq Select transcripts are integral to the MANE (Matched Annotation from NCBI and EMBL-EBI) project. See The MANE project section for details.
The next three sections describe the process involved in choosing the RefSeq Select transcript in eukaryotic annotations.
Picking the RefSeq Select transcript
NCBI has developed a pipeline that picks the RefSeq Select transcript based on multiple hierarchically scored criteria. Some of the criteria are specific to the human genome, for example, the prior assignment of the transcript as a ‘Reference Standard’ in the RefSeqGene set for a gene that is also included in the public LRG (Locus Reference genomic) set. Other criteria such as expression score, which is computed from RNA-seq intron-spanning alignments, apply to all taxa in scope. Figure 1 provides a simplified outline of the RefSeq Select pipeline. The computational pipeline is complemented by input and QA from expert curators in the RefSeq group who help maintain the quality of the RefSeq Select transcripts and make the transcript choice in complex loci and other genes where the pipeline choice may not be ideal.
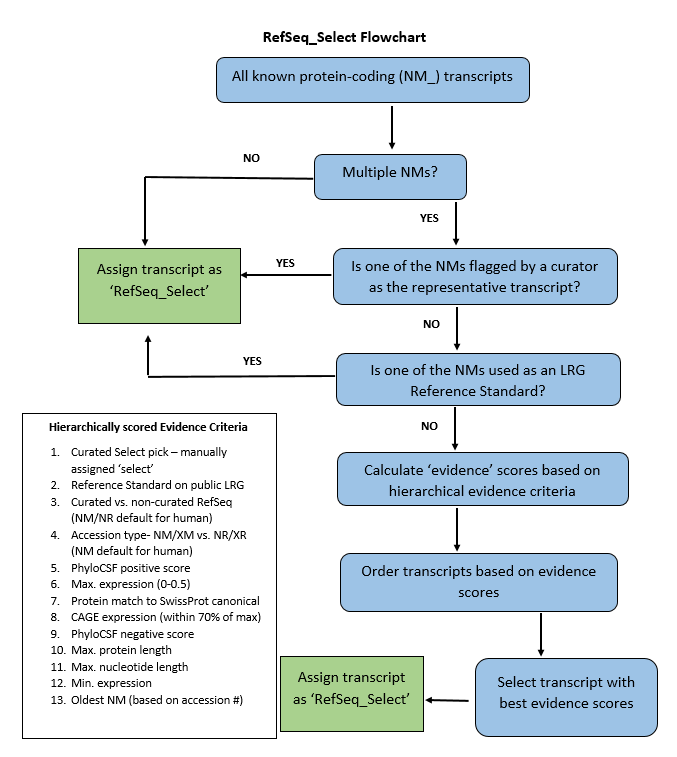
Figure 1. Workflow for choosing the RefSeq Select transcript
Description of RefSeq Select criteria (based on the human RefSeq Select set)
- Curated RefSeq Select pick: If a curator determines a known RefSeq to be the best suited transcript for the RefSeq Select set, the transcript will be picked by the Select pipeline and override all other criteria.
- Prior use as a clinical standard: If a known RefSeq is already in use as a reference transcript in a public LRG record, it will be the default RefSeq Select choice. If multiple RefSeqs qualify for this criterion, then the other Select criteria will be applied to only the qualifying transcripts, to pick the best among them.
- Curated (NM_, NP_, NR_) versus non-curated (XM_, XP_, XR_) RefSeqs: In the human and mouse RefSeq Select set, curated (or ‘known’) RefSeqs (see About RefSeq) are chosen by default.
- Accession type (NM_/XM_ versus NR_/XR_): For protein-coding genes, preference is given to coding (NM_, XM_) non-coding (NR_, XR_) RefSeqs.
- Conservation of the coding region of the transcript: Evolutionary conservation of the coding region is calculated based on PhyloCSF data. PhyloCSF is a method that determines the protein-coding potential of individual bases using alignments of the coding regions of multiple organisms representing a range of taxonomic groups. PhyloCSF scores are calculated based on codon substitution frequencies. Positive PhyloCSF scores indicate conservation of the nucleotides in the coding region (CDS). Preference is given to transcripts with more positive scoring bases in the CDS, treating transcripts with similar scores (within 90-bp of the maximum) as equivalent.
- Transcript expression: A composite expression score is calculated for each transcript based on ‘read scores’ (number of short-read RNA-seq sequences spanning the intron, also referred to as ‘split reads’) of individual introns, which are based on the combination of short-read RNA-seq studies used in the RefSeq annotation and available long-read data. The score penalizes introns that are under-represented compared to their neighbors and favors more splices as a proxy for favoring full-length transcripts. Transcripts with similar expression scores are considered equally expressed.
- Protein match to the Swiss-Prot canonical: Transcripts that encode a protein that matches the Swiss-Prot canonical isoform.
- CAGE expression: This criterion is applicable to genes that produce transcripts from different transcription start sites (TSSs) or promoters. Expression levels of transcripts are indicated by a high-throughput sequencing technique called Cap Analysis of Gene Expression (CAGE), which produces a genome-wide snapshot of 5’ ends of the mRNA pool from a biological sample. The RefSeq Select pipeline leverages RefSeq-processed CAGE clusters, which are calculated from the CAGE cluster and TSS data available from the FANTOM consortium. Transcripts associated with the CAGE cluster with the highest CAGE score (total tag counts), and those associated with CAGE clusters that have a score that is within 70% of the maximum CAGE score, are considered equally expressed.
- PhyloCSF negative score: A negative PhyloCSF score may indicate non-conserved CDS bases. The protein-coding transcript with the least negative score is preferred over other transcripts. Note: PhyloCSF has limitations in certain situations, for example very short exons. Such cases are deferred to manual review.
- Maximum protein length: This criterion picks the transcript(s) encoding the longest protein.
- Nucleotide length: This criterion picks the longest transcript.
- Minimum transcript expression: The transcript with the least expression score of all the transcripts associated with a gene.
- Oldest accession: This criterion is meant to act as a ‘tie-breaker’ when multiple transcripts have the same scores for all the above listed criteria.
How are the transcripts scored?
Every transcript gets a binary score for each of the above criteria. The set of alternative transcripts for a gene are then analyzed hierarchically to identify a single transcript that scores better than the others. For example, if all coding transcripts have CDSes with similar PhyloCSF scores (criteria #5), but one has notably better expression (criteria #6), then that transcript is chosen.
How can the RefSeq Select transcript be accessed?
Depending on which NCBI resource one is looking at, there are multiple markups to distinguish the RefSeq Select transcript from other transcripts of a gene.
NCBI gene summary box
A gene summary box is returned in response to organism-gene queries (e.g. “mouse Igf1”) on the NCBI homepage and NCBI sequence databases. The gene summary box includes expandable ‘RefSeq transcripts’ and ‘RefSeq proteins’ sections that list up to five curated RefSeqs. The RefSeq Select transcript or protein is labeled and sorted to the top of this list (Figure 2).
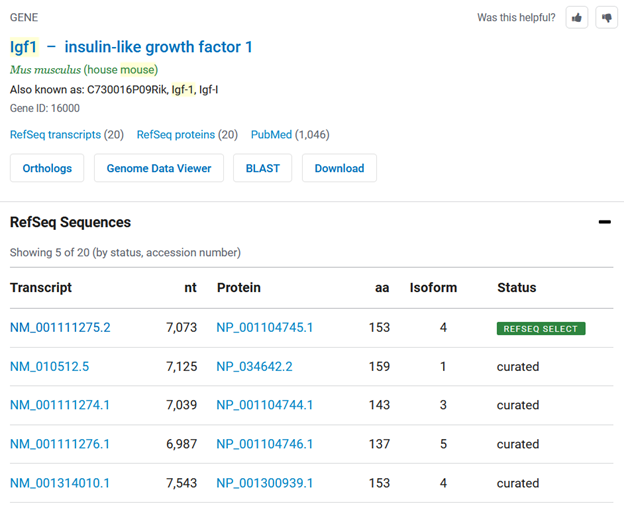
Figure 2. The search results gene summary page (with the transcripts tab expanded) showing the RefSeq Select markup.
RefSeq flatfile keyword
The RefSeq flatfile contains ‘RefSeq Select’ in the Keyword section (Figure 3). In the case of human RefSeq Select transcripts, ‘MANE Select’ may appear instead of ‘RefSeq Select’.‘MANE Select’ is a subset of the human ‘RefSeq Select’ dataset being developed in coordination with EMBL-EBI (see The MANE project section below).

Figure 3. A portion of the flatfile view of NM_001111275.2 (mouse gene Igf1), showing the ‘RefSeq Select’ markup (underlined in the screenshot) in the keyword section.
RefSeq flatfile attribute
The RefSeq flatfile of the RefSeq Select transcript contains an attribute section in which the ‘Refseq Select criteria’ attribute lists the main criteria based on which the transcript was chosen (Figure 4)

Figure 4. A portion of the flatfile view of NM_001111275.2, showing the ‘RefSeq Select criteria’ attribute in the ‘RefSeq Attributes’ section, with the selection criteria that led to the choice of this transcript as RefSeq Select for the mouse gene Igf1.
RefSeq Select criteria can be one or more of the following:
- manual assertion: the transcript has been manually chosen as RefSeq select based on the combined evidence available for the gene
- conservation: the transcript is comparable to or better than other RefSeq transcripts based on PhyloCSF positive bases in the CDS, and within 90-bp of the maximum observed for the gene (Selection criterion #5)
- expression: the transcript is comparable to or better than other RefSeq transcripts based on intron splicing data (Selection criterion #6)
- longest protein: the protein is as long as, or longer than all other RefSeq proteins for the gene (Selection criterion #10)
- single protein-coding transcript: the transcript is the only protein-coding transcript currently annotated for the gene
- computational evidence: the selection is based on criteria other than the above
Note: Other criteria described in the Selection Criteria section above may also affect the transcript choice for RefSeq Select but aren’t currently reported on the flatfile.
NCBI Entrez search
RefSeq Select accessions can be searched in the Nucleotide database by using the Entrez RefSeq Select filter. For example, “PALM[gene] AND Refseq_select[filter]” will return the nucleotide record of NM_002579.3, the RefSeq Select transcript of this gene. The entire list of human RefSeq Select accessions, including the subset in the ‘MANE Select’ dataset, can be extracted using the Entrez query "Homo sapiens"[Organism] AND Refseq_select[filter]”. The list can then be downloaded and saved to a file using the “Send to” tab at the top of the Nucleotide results page.
RefSeq annotation files
The annotation is available via FTP. Column 9 in the GFF and GTF files contain a “RefSeq Select” or “MANE Select” tag attribute (tag=MANE Select in GFF3, or tag ”MANE Select” in GTF) in the rows associated with the mRNA, CDS and exon features.
Using RefSeq Select
The RefSeq Select set is a set of representative transcripts that are well-supported by experimental data and are meant to represent the biology of the gene, using proxies such as transcript expression levels and the evolutionary conservation of the coding region. In selecting the transcript, we also try to synchronize the RefSeq Select with data in other databases that represent one or more representative/canonical forms, for example, the Swiss-Prot canonical isoform and the reference transcript in the Locus Reference Genomic (LRG) dataset. The RefSeq Select transcript, therefore, is meant to serve as a representative transcript of a gene in studies and applications, such as evolutionary analyses, comparative genomics and clinical variant reporting, that may require the use of only one transcript per gene. It eliminates the need for users to apply their own criteria (which may be inconsistent among different users) to pick a representative transcript. As a cautionary note, the RefSeq Select set is recommended for applications that may require a single transcript per gene; but it does not diminish the importance of the remaining transcripts and proteins. If users need to get a comprehensive picture of the transcriptional diversity of a gene, the entire set of RefSeq transcripts and proteins should be examined.
The MANE project
In 2018, NCBI and EMBL-EBI (European Molecular Biology Laboratories-European Bioinformatics Institute) announced a new collaborative project called Matched Annotation from NCBI and EMBL-EBI (MANE). This project aims to provide a matched set of transcripts for every human protein-coding gene. The transcripts in this set are annotated identically in the RefSeq and the Ensembl-GENCODE gene sets. As a first step in the project the MANE Select set is now available, which consists of a single representative or “Select” transcript for each human protein-coding gene. Currently, the MANE Select is a beta set that covers over 80% of the protein-coding genes. NCBI and EBI will add to the set in the next year with the goal of achieving close to 100% coverage of protein-coding genes. Details of the MANE project are available here.
Relationship between RefSeq Select and MANE Select
The MANE Select dataset is a subset of RefSeq Select. For a given human protein-coding gene, the RefSeq Select transcript is designated as the ‘MANE Select’ when it matches the Ensembl ‘Select’ transcript and is included in the public MANE set. In the case of MANE Select transcripts, the keyword ‘RefSeq Select’ is replaced by ‘MANE Select’ in the RefSeq flatfile (Figure 5).

Figure 5. A portion of the flatfile view of NM_012384.5 (gene GMEB2), showing the ‘MANE Select’ markup in the keyword section
In addition, an attribute, ‘MANE Ensembl match’, in the RefSeq flatfile provides the identifiers of the matching Ensembl transcript and protein (Figure 6.

Figure 6. A portion of the flatfile view of NM_001238.4 (gene CCNE1), showing the ‘MANE Ensembl match’ attribute with the matching Ensembl transcript and protein identifiers.
Feedback
We welcome your feedback on the RefSeq Select project. Please use the yellow vertical Feedback tab on the bottom right of the page to send us your comments and suggestions.