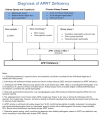
Clinical Description
More than 400 individuals with adenine phosphoribosyltransferase (APRT) deficiency have been reported in the medical literature [Bollée et al 2010, Bollée et al 2012, Harambat et al 2012, Balasubramaniam et al 2014, Zaidan et al 2014, Runolfsdottir et al 2016, Huq et al 2018, Runolfsdottir et al 2019a, Runolfsdottir et al 2019b].
Table 2.
Presenting Renal Manifestations in APRT Deficiency
View in own window
| Presenting Renal Manifestation | Approximate Frequency |
|---|
| Kidney stone disease 1 | 60%-90% |
| Chronic kidney disease 2, 3 | >50% |
| Acute kidney injury 4 | 30% 5 |
| End-stage renal disease | 15% |
- 1.
In both children and adults
- 2.
- 3.
Due to DHA crystal nephropathy
- 4.
Due to urinary tract obstruction
- 5.
Age at presentation. APRT deficiency may present at any age; there is no typical age of clinical onset. However, in at least 50% of affected individuals, symptoms do not occur until adulthood.
The age at diagnosis among individuals in the APRT Deficiency Registry of the Rare Kidney Stone Consortium (RKSC) ranged from six months to 72 years (median age: 37 years) [
Runolfsdottir et al 2016].
Approximately 35% of persons with APRT deficiency are diagnosed before age 18 years.
In a significant number of asymptomatic individuals, a diagnosis of APRT deficiency has been suggested by the detection of DHA crystals on routine urine microscopy or through the screening of sibs of affected individuals and subsequently confirmed by enzyme activity or genetic testing.
Of note, abdominal ultrasound and CT examinations performed for other reasons may identify kidney stones in individuals with APRT deficiency who may be otherwise asymptomatic [Huq et al 2018].
Kidney stone disease. Between 60% and 90% of affected individuals have already developed kidney stones at diagnosis. In the absence of pharmacotherapy (see Management,
Table 4), the majority of those untreated symptomatic persons experience stone recurrence [Runolfsdottir et al 2019a], abdominal pain, and/or lower urinary tract symptoms (e.g., straining, hesitancy, dribbling, incomplete bladder emptying).
Chronic kidney disease (CKD) secondary to DHA crystal nephropathy is present in more than 50% of individuals at diagnosis [Runolfsdottir et al 2016]. As many as 15% of affected individuals have progressed to end-stage renal disease (ESRD) at the time of diagnosis of APRT deficiency [Harambat et al 2012, Runolfsdottir et al 2016].
ESRD. Approximately 20%-25% of affected individuals develop ESRD, usually in adult life, if adequate treatment is not provided.
APRT deficiency is not known to affect organs other than the kidney; however, the authors and other investigators have encountered occasional individuals with APRT deficiency complaining of eye discomfort [Neetens et al 1986; Author, personal observation], which merits further study.
Nomenclature
Originally, two types of APRT deficiency with identical clinical manifestations were described, based on the level of residual APRT activity in cell extracts (erythrocyte lysates) [Sahota et al 2001]. However, this distinction is of historical interest only, as APRT enzyme activity in intact cells has been shown to be less than 1% in both types [Kamatani et al 1985] (see APRT Enzyme Activity).
Prevalence
The estimated heterozygote frequency in different populations ranges from 0.4% to 1.2% [Hidaka et al 1987, Sahota et al 2001], suggesting that the prevalence of a homozygous state is at least 1:50,000 to 1:100,000.
If this holds true, at least 70,000-80,000 individuals should be affected worldwide, of whom 40,000 would be expected to be in Asia, 9,000 in Europe, and 8,000 in the Americas, including at least 3,000 affected individuals in the US alone. Most of these individuals are currently unrecognized, and thus not benefitting from medical therapy.
Evidence suggests that APRT deficiency may be a seriously underrecognized cause of kidney stones and crystal nephropathy, progressing over time to ESRD in a significant proportion of untreated individuals [Zaidan et al 2014].