Clinical Description
The clinical course of polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL) can be divided into four stages: latent, osseous, early neurologic, and late neurologic [Klünemann et al 2005].
Latent stage. Early development is normal.
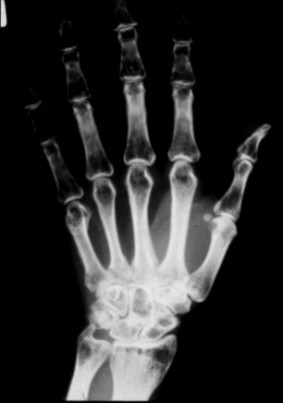
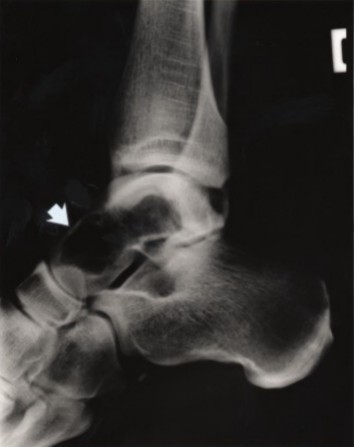
Osseous stage (3rd decade of life). The first symptoms of PLOSL appear in early adulthood as pain and tenderness, mostly in the ankles and feet, usually following strain or a minor accident. Fractures are typically diagnosed several years later, most commonly in the bones of the extremities [Paloneva et al 2001]. The first fractures usually occur shortly before age 30 years; however, affected individuals may have had pain and swelling of the ankles and wrists after strain for years. The fractures heal well. It is important to note that some individuals may present with neurologic symptoms without any preceding osseous manifestations [Paloneva et al 2001, Bock et al 2013].
Early neurologic stage (4th decade of life). Personality changes begin insidiously in the fourth decade. Affected individuals show progressive loss of judgment, leading to serious social consequences, including divorce, unemployment, and financial trouble [Paloneva et al 2001, Ilonen et al 2012]. Some individuals may attempt suicide. The full-blown picture of frontal lobe syndrome subsequently appears: loss of judgment; euphoria; lack of social inhibitions, including Witzelsucht; disturbance of concentration; and lack of insight, libido, and motor persistence.
Progressive signs of upper motor neuron involvement (spasticity, extensor plantar reflexes) are noticed. With advancing disease, lack of initiative and activity conceal the aforementioned symptoms [Paloneva et al 2001].
Memory disturbances begin at approximately the same age as the personality changes, and are best detectable by psychometric tests [Vanhanen et al 2013]. The memory disturbance is less severe than the personality change, and affected individuals retain basic personal information until the last stage of the disease.
Other disturbances of higher cortical function, such as motor aphasia, agraphia, acalculia, and apraxia, appear only at the last stage of the disease.
Affected individuals may develop postural dyspraxia: they walk or sit in peculiar skewed postures. Involuntary athetotic or choreatic movements or myoclonic twitches are common. Individuals who reach their mid-thirties frequently experience epileptic seizures. In some individuals, impotence or lack of libido and urinary incontinence are among the first symptoms [Paloneva et al 2001].
Late neurologic stage. In the last stage of the disease, individuals lose their ability to walk and progress to a vegetative state. Primitive reflexes, such as visual and tactile grasp and mouth-opening reflexes, as well as the sucking reflex, may become noticeable. Affected individuals typically die before age 50 years [Paloneva et al 2001].
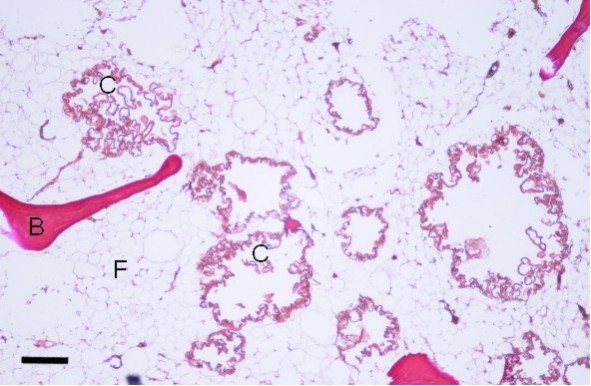
Bone pathology. The cyst-like bone lesions are filled with lipid material that microscopically consists of characteristic 1-2 µm thick lipid membranes and amorphous lipid substance [Kitajima et al 1989]. See .
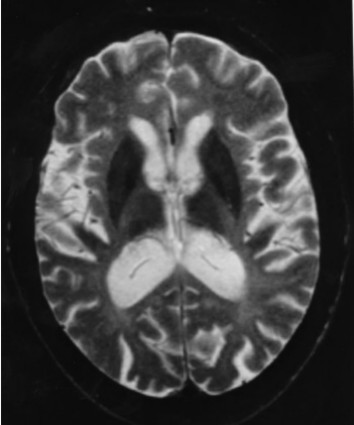
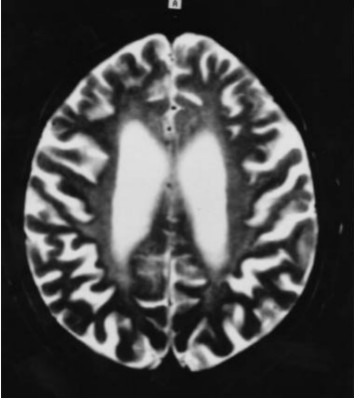
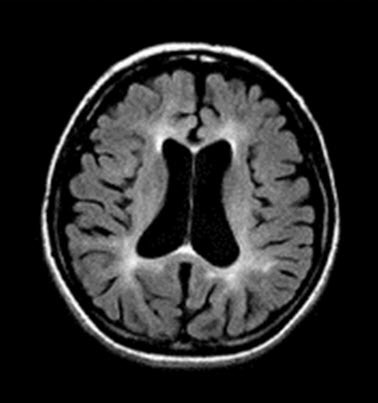
Neuropathology. Generalized cerebral gyral atrophy with frontal accentuation is observed at autopsy. The corpus callosum is abnormally thin. The central white matter is severely reduced in amount, grayish, and tough. The basal ganglia, particularly the caudate nuclei, are variably reduced in size [Paloneva et al 2001]. All affected individuals show marked hydrocephalus ex vacuo.
Histologic examination reveals scattered neurons showing features of central chromatolysis. Intraneuronal or glial pathologic inclusions have not been observed [Paloneva et al 2001]. Neuronal loss as well as astrocytic proliferation and hypertrophy are observed in the caudate nuclei. In addition, scattered calcospherites are seen, particularly in the putamina and globi pallidi [Kalimo et al 1994, Paloneva et al 2001]. Thalamic degeneration may occur [Kobayashi et al 2000]. Affected individuals show advanced loss of axons and myelin and a pronounced astrocytic reaction in the centrum semiovale, accentuated in the frontal and temporal lobes, with moderate involvement of the gyral white matter. In addition, widespread activation of microglia in the cerebral white matter is seen [Paloneva et al 2001]. Scattered small arterioles and capillaries in the deep frontal and temporal white matter show concentric thickening of the vascular wall with multiple thickened basement membranes and narrowing or obliteration of the lumen [Paloneva et al 2001]. The cerebral cortices are less severely affected [Aoki et al 2011].
Pathologic findings in other organs. Characteristic lipomembranous changes have been described in systemic adipose tissue [Nasu et al 1973]. Pathologic manifestations in organs other than the central nervous system (CNS) and the skeletal system have been insufficiently characterized.