Classic (Severe) RCDP1
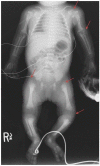
The characteristic clinical features of classic RCDP1 are skeletal abnormalities, cataracts, growth restriction, and intellectual disability. Life expectancy is shortened: the majority of children do not survive beyond the first decade of life and a proportion die in the neonatal period. Of 35 affected children older than age one month, 90% survived to age one year, 55% to age five years, and approximately 20% to age 12 years [White et al 2003]. In a separate review of 66 individuals with RCDP1, 80% survived to age five years, 45% to age 12 years, and 35% to adulthood [Duker et al 2020]. Most deaths in these cohorts were secondary to respiratory complications. Some infants may die in the neonatal period; this number is not known. Clinical experience suggests that neonatal deaths were associated with congenital heart disease or lung hypoplasia [Oswald et al 2011].
Skeletal findings. Infants with RCDP1 have bilateral shortening of the humerus and to a lesser degree the femur. They typically have contractures and stiff, painful joints, causing irritability in infancy.
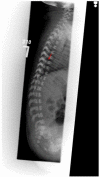
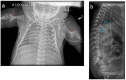
In a study of the MRIs of children with classic RCDP1, all individuals had cervical stenosis. More global spinal stenosis, cervical kyphosis, and thoracolumbar kyphosis were seen, but with less frequency. Tethered cord was also identified [Khanna et al 2001, Bams-Mengerink et al 2013, Abousamra et al 2019].
Cataracts. Bilateral cortical cataracts develop in virtually all affected individuals. They are usually present at birth or appear in the first few months of life and are progressive.
Growth restriction. Whereas birth weight, length, and head circumference are often at the lower range of normal, profound postnatal growth deficiency is evident throughout the life span [Duker et al 2017]. At age three years, height, weight and head circumference are around the 50th percentile for a child age 4-6 months. Rate of weight gain is slow, dropping to 5 g/day at age six months, and <2 g/day expected after age three years. RCDP1 height, weight, and head circumference growth charts are available as well as height-for-weight charts and charts showing rate of weight gain over time [Duker et al 2017].
Intellectual disability. Developmental quotients are below 30. Early developmental skills such as smiling and recognizing voices are achieved by most children with RCDP1, but at delayed ages. Skills achieved in typically developing children after age six months are never seen in children with RCDP1 [White et al 2003, Bams-Mengerink et al 2013].
Seizures. The majority of children develop seizures [White et al 2003, Bams-Mengerink et al 2013]. Myoclonic jerks are the most frequent type of seizure reported, but seizure frequency and types are variable. The median age at seizure onset was 2.5 years [Bams-Mengerink et al 2013].
Recurrent respiratory tract infections. Most children with RCDP1 have recurrent respiratory tract infections caused by a combination of neurologic compromise, aspiration, immobility, and a small chest with restricted expansion. Plasmalogen deficiency may also play a role in the chronic respiratory disease as these lipids are enriched in lung tissues and an integral component of surfactant [Oswald et al 2011, Braverman & Moser 2012, Duker et al 2020].
Congenital heart disease. Cardiac malformations have been identified in 52% and 64% of individuals with RCDP1 in the Dutch and North American cohorts respectively [Huffnagel et al 2013, Duker et al 2016]. Septal defects, tetralogy of Fallot, and peripheral pulmonary stenosis were most commonly reported. Mitral valve prolapse was also noted in several individuals.
Other. Eczema, mild ichthyosis, and skin rashes were noted in around 50% of individuals in the cohort studied by White et al [2003].
Other malformations observed in one affected individual include: ureteropelvic junction (UPJ) obstruction [Khanna et al 2001], cleft palate, diaphragmatic hernia, hypospadias, and cryptorchidism [White et al 2003].
Routine brain imaging is normal or shows cerebral and cerebellar atrophy with enlargement of the ventricles and CSF spaces [Powers et al 1999]. Cerebellar atrophy is progressive [Bams-Mengerink et al 2006]. MRI and MR spectroscopy have shown delayed myelinization, signal abnormalities in supratentorial white matter, decreased choline-to-creatine ratios, and increased levels of mobile lipids, thought to reflect the deficiency of plasmalogens, which are substantial components of myelin [Alkan et al 2003, Bams-Mengerink et al 2006, Bams-Mengerink et al 2013].
Nonclassic (Mild) RCDP1
This group is defined clinically by the ability to walk with or without support and the ability to use verbal or nonverbal types of communication [Bams-Mengerink et al 2013]. The majority of individuals with nonclassic (mild) RCDP1 have presented in early childhood with bilateral cataracts, multiple joint contractures, and developmental delays [Braverman et al 2002, Bams-Mengerink et al 2006]. A few individuals manifest cataracts within the first two years, no skeletal findings, and behavioral disorders that develop at school age [Braverman et al 2002, Yu et al 2013]. Overall life expectancy is considerably longer than that of classic RCDP1, with survival to adulthood [Bams-Mengerink et al 2013, Huffnagel et al 2013]; in a recent study 11 of 12 affected individuals survived to adulthood [Duker et al 2020].
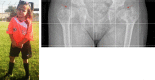
Skeletal. Most individuals with mild RCDP1 have limited joint mobility due to flexion contractures of the elbows, knees, and hips. While the radiographic finding of CDP (chondrodysplasia punctata) is commonly noted at the time of RCDP1 diagnosis, rhizomelic limb shortening is uncommon in this group. Deformities in hip joints including coxa vara and small femoral heads have been reported (see ) [Barth et al 1996, Bams-Mengerink et al 2013]. Many of these individuals required orthopedic surgeries over time to improve mobility and activities of daily living [Barth et al 1996].
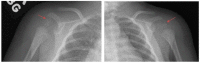
Girl age five years with nonclassic RCDP1 showing better growth than classic RCDP1 and absence of rhizomelia. X-rays of the hips show coxa vara of the femoral head (arrows). Femoral osteotomy was performed to improve mobility. All images provided by authors. (more...)
Cataract. Most individuals with mild RCDP1 have bilateral cataracts diagnosed in the first two years of life.
Growth of individuals with mild RCDP1 can be within normal ranges at birth. Postnatal growth rates are also better than those in classic RCDP1. Growth curves based on four individuals with nonclassic RCDP1 have been published [Duker et al 2017].
Intellect. Most individuals with mild RCDP1 have had developmental delays and learning disabilities. However, they were able to achieve gross and fine motor skills never achieved in individuals with classic RCDP1. All individuals with mild RCDP1 were able to walk and most can communicate verbally; all required some degree of special education. Brain MRI was normal in all three individuals with nonclassic RCDP1 reported by Bams-Mengerink et al [2006].
Seizures. In one cohort, three of four individuals with mild RCDP1 developed seizures in late childhood (age range 7-21 years). The type of seizures more commonly seen in this group were absence and tonic-clonic seizures [Bams-Mengerink et al 2013].
Congenital heart disease. In six individuals with mild RCDP1, cardiac defects including atrial septal defects were reported in two individuals, one of whom also had first-degree heart block. Two individuals developed mitral valve prolapse, possibly indicating degenerative cardiac changes [Huffnagel et al 2013].
Behavior disorders, usually identified at school age in the limited number of individuals described with mild RCDP1, included attention-deficit/hyperactivity disorder and autism spectrum disorder (ASD) [Bams-Mengerink et al 2013, Yu et al 2013].
In one family, two sibs with the PEX7 variant p.Ser25Phe had congenital cataract. One sib had ASD, intellectual disability, and epilepsy; the other had normal intellect with attention-deficit disorder. Elevated blood phytanic acid was observed on an unrestricted diet.
In another family, two sibs had the PEX7 variant p.Trp75Cys and ASD, intellectual disability, epilepsy, and cataracts.
Retinitis pigmentosa and peripheral neuropathy. An individual with the PEX7 variant c.-45C>T had developmental delays and poor growth in childhood; retinitis pigmentosa and peripheral neuropathy developed in adolescence [Braverman et al 2002]. Cataracts were not reported.