Summary
Clinical characteristics.
Rhizomelic chondrodysplasia punctata type 1 (RCDP1), a peroxisome biogenesis disorder (PBD) has a classic (severe) form and a nonclassic (mild) form. Classic (severe) RCDP1 is characterized by proximal shortening of the humerus (rhizomelia) and to a lesser degree the femur, punctate calcifications in cartilage with epiphyseal and metaphyseal abnormalities (chondrodysplasia punctata, or CDP), coronal clefts of the vertebral bodies, and cataracts that are usually present at birth or appear in the first few months of life. Birth weight, length, and head circumference are often at the lower range of normal; postnatal growth deficiency is profound. Intellectual disability is severe, and the majority of children develop seizures. Most affected children do not survive the first decade of life; a proportion die in the neonatal period. Nonclassic (mild) RCDP1 is characterized by congenital or childhood cataracts, CDP or infrequently, chondrodysplasia manifesting only as mild epiphyseal changes, variable rhizomelia, and milder intellectual disability and growth restriction than classic RCDP1.
Diagnosis/testing.
The diagnosis of RCDP1 is established in a proband with suggestive clinical, radiographic, and laboratory findings and biallelic pathogenic variants in PEX7 identified on molecular genetic testing.
Management.
Treatment of manifestations: Classic (severe) RCDP1: Management is supportive and limited by the multiple handicaps present at birth and poor outcome. Poor feeding and recurrent aspiration may necessitate placement of a gastrostomy tube; attention to respiratory function and good pulmonary toilet. Cataract extraction may restore some vision. Physical therapy to improve contractures; orthopedic procedures may improve function in some individuals. Management of developmental delay/intellectual disability as per standard of care.
Prevention of primary manifestations: Dietary restriction of phytanic acid to avoid the consequences of phytanic acid accumulation over time may benefit individuals with mild RCDP1.
Surveillance: Frequent monitoring of growth, nutritional status, and developmental and educational needs; regular assessments for evidence of aspiration, respiratory insufficiency, seizure control, vision, hearing, contractures, and orthopedic complications.
Genetic counseling.
RCDP1 is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a PEX7 pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting both pathogenic variants and being affected, a 50% chance of inheriting one pathogenic variant and being an unaffected carrier, and a 25% chance of inheriting both normal alleles. Molecular genetic carrier testing of at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible once the PEX7 pathogenic variants have been identified in an affected family member.
Diagnosis
Suggestive Findings
Rhizomelic chondrodysplasia punctata type 1 (RCDP1) should be suspected based on the individual's age and the severity of clinical and skeletal/radiographic findings.
Classic (Severe) RCDP1 (the majority of affected individuals)
Neonatal period
- Congenital cataracts
- Skeletal/radiographic findings
- Rhizomelia (proximal shortening of the long bones)
- Chondrodysplasia punctata (CDP). Punctate calcifications observed in radiographs in the epiphyseal cartilage at the knee, hip, elbow, and shoulder that can be more extensive, involving the hyoid bone, larynx, costochondral junctions, and vertebrae. Metaphyseal abnormalities may be present (see Figure 1).
- Radiolucent coronal clefts of the vertebral bodies on lateral spine radiographs that represent unossified cartilage (See Figure 2.)
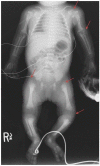
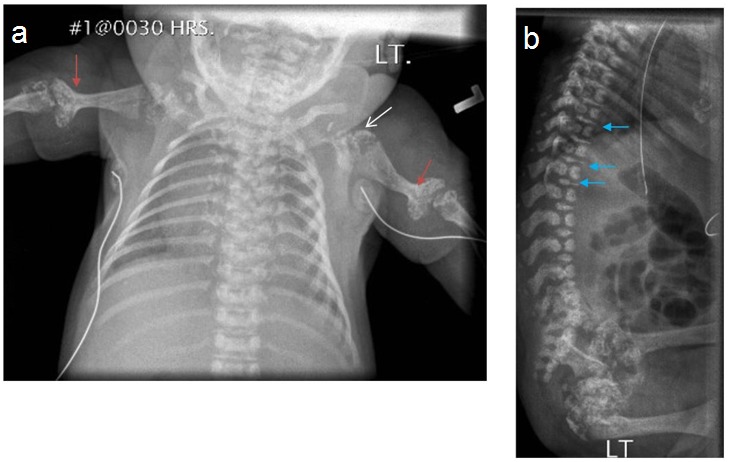
Figure 1.
X-ray of a newborn with classic (severe) RCDP showing rhizomelia and chondrodysplasia punctata (CDP) (arrows).
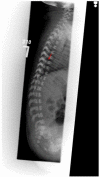
Figure 2.
Lateral Infant spine showing vertebral coronal clefting (arrow)
Childhood
- Cataracts (usually apparent by age 6 months)
- Severe intellectual disability
- Profound postnatal growth restriction
- Evolution of skeletal/radiographic findings:
- Resolution of the punctate calcifications leaving abnormal epiphyses and flared and irregular metaphyses after ages one to three years (See Figure 3a.)
- Possible calcification of the intervertebral discs (See Figure 3b.)
- Frontal bossing and a short, concave nasal bridge resulting from cartilaginous involvement (See Figure 4: infant and older children with classic RCDP1.)
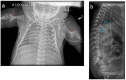
Figure 3
a. Progression of radiographic changes in a child with classic (severe) RCDP1 showing metaphyseal flaring (red closed arrows), epiphyseal abnormalities (white open arrow) b. Calcification of intervertebral discs (blue arrows)

Nonclassic (Mild) RCDP1
- Congenital or childhood cataracts
- Chondrodysplasia punctata (CDP) or infrequently, chondrodysplasia manifesting as mild epiphyseal changes only (See Figure 5.)
- Variable rhizomelia
- Milder intellectual disability and growth restriction than classic RCDP1
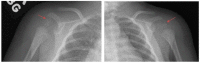
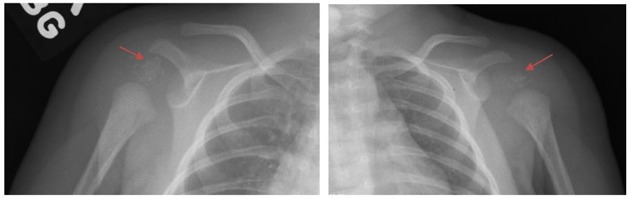
Figure 5.
X-rays of nonclassic (mild) RCDP1: Shoulders show CDP (arrows) at the proximal humerus without metaphyseal changes or rhizomelia.
Biochemical Testing
The finding of deficiency of plasmalogens in red blood cells, increased plasma concentration of phytanic acid (when diet includes phytanic acid sources), and normal plasma concentration of very long chain fatty acids (VLCFA) has consistently predicted the PEX7 receptor defect in RCDP1 [Braverman et al 2002, Motley et al 2002]. These assays are extremely specialized and are reliably performed in a limited number of laboratories worldwide.
Plasmalogen levels are valuable in distinguishing between classic and milder RCDP1: erythrocyte plasmalogen levels are around 10- to 30-fold higher in milder RCDP1 than in classic RCDP1 [Braverman et al 2002, Bams-Mengerink et al 2013, Duker et al 2016].
Establishing the Diagnosis
The diagnosis of RCDP1 is established in a proband with suggestive findings and biallelic pathogenic (or likely pathogenic) variants in PEX7 identified by molecular genetic testing (see Table 1).
Note: Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. Identification of biallelic PEX7 variants of uncertain significance (or of one known PEX7 pathogenic variant and one PEX7 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing). Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings of classic RCDP1 described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with the less specific findings of nonclassic (mild) RCDP1 are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
A chondrodysplasia punctata multigene panel that includes PEX7 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 1).
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the diagnosis of RCDP1 is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is an option. Exome sequencing is the most commonly used genomic testing method; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis. For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Rhizomelic Chondrodysplasia Punctata Type 1
Clinical Characteristics
Clinical Description
Classic (Severe) RCDP1
The characteristic clinical features of classic RCDP1 are skeletal abnormalities, cataracts, growth restriction, and intellectual disability. Life expectancy is shortened: the majority of children do not survive beyond the first decade of life and a proportion die in the neonatal period. Of 35 affected children older than age one month, 90% survived to age one year, 55% to age five years, and approximately 20% to age 12 years [White et al 2003]. In a separate review of 66 individuals with RCDP1, 80% survived to age five years, 45% to age 12 years, and 35% to adulthood [Duker et al 2020]. Most deaths in these cohorts were secondary to respiratory complications. Some infants may die in the neonatal period; this number is not known. Clinical experience suggests that neonatal deaths were associated with congenital heart disease or lung hypoplasia [Oswald et al 2011].
Skeletal findings. Infants with RCDP1 have bilateral shortening of the humerus and to a lesser degree the femur. They typically have contractures and stiff, painful joints, causing irritability in infancy.
In a study of the MRIs of children with classic RCDP1, all individuals had cervical stenosis. More global spinal stenosis, cervical kyphosis, and thoracolumbar kyphosis were seen, but with less frequency. Tethered cord was also identified [Khanna et al 2001, Bams-Mengerink et al 2013, Abousamra et al 2019].
Cataracts. Bilateral cortical cataracts develop in virtually all affected individuals. They are usually present at birth or appear in the first few months of life and are progressive.
Growth restriction. Whereas birth weight, length, and head circumference are often at the lower range of normal, profound postnatal growth deficiency is evident throughout the life span [Duker et al 2017]. At age three years, height, weight and head circumference are around the 50th percentile for a child age 4-6 months. Rate of weight gain is slow, dropping to 5 g/day at age six months, and <2 g/day expected after age three years. RCDP1 height, weight, and head circumference growth charts are available as well as height-for-weight charts and charts showing rate of weight gain over time [Duker et al 2017].
Intellectual disability. Developmental quotients are below 30. Early developmental skills such as smiling and recognizing voices are achieved by most children with RCDP1, but at delayed ages. Skills achieved in typically developing children after age six months are never seen in children with RCDP1 [White et al 2003, Bams-Mengerink et al 2013].
Seizures. The majority of children develop seizures [White et al 2003, Bams-Mengerink et al 2013]. Myoclonic jerks are the most frequent type of seizure reported, but seizure frequency and types are variable. The median age at seizure onset was 2.5 years [Bams-Mengerink et al 2013].
Recurrent respiratory tract infections. Most children with RCDP1 have recurrent respiratory tract infections caused by a combination of neurologic compromise, aspiration, immobility, and a small chest with restricted expansion. Plasmalogen deficiency may also play a role in the chronic respiratory disease as these lipids are enriched in lung tissues and an integral component of surfactant [Oswald et al 2011, Braverman & Moser 2012, Duker et al 2020].
Congenital heart disease. Cardiac malformations have been identified in 52% and 64% of individuals with RCDP1 in the Dutch and North American cohorts respectively [Huffnagel et al 2013, Duker et al 2016]. Septal defects, tetralogy of Fallot, and peripheral pulmonary stenosis were most commonly reported. Mitral valve prolapse was also noted in several individuals.
Other. Eczema, mild ichthyosis, and skin rashes were noted in around 50% of individuals in the cohort studied by White et al [2003].
Other malformations observed in one affected individual include: ureteropelvic junction (UPJ) obstruction [Khanna et al 2001], cleft palate, diaphragmatic hernia, hypospadias, and cryptorchidism [White et al 2003].
Routine brain imaging is normal or shows cerebral and cerebellar atrophy with enlargement of the ventricles and CSF spaces [Powers et al 1999]. Cerebellar atrophy is progressive [Bams-Mengerink et al 2006]. MRI and MR spectroscopy have shown delayed myelinization, signal abnormalities in supratentorial white matter, decreased choline-to-creatine ratios, and increased levels of mobile lipids, thought to reflect the deficiency of plasmalogens, which are substantial components of myelin [Alkan et al 2003, Bams-Mengerink et al 2006, Bams-Mengerink et al 2013].
Nonclassic (Mild) RCDP1
This group is defined clinically by the ability to walk with or without support and the ability to use verbal or nonverbal types of communication [Bams-Mengerink et al 2013]. The majority of individuals with nonclassic (mild) RCDP1 have presented in early childhood with bilateral cataracts, multiple joint contractures, and developmental delays [Braverman et al 2002, Bams-Mengerink et al 2006]. A few individuals manifest cataracts within the first two years, no skeletal findings, and behavioral disorders that develop at school age [Braverman et al 2002, Yu et al 2013]. Overall life expectancy is considerably longer than that of classic RCDP1, with survival to adulthood [Bams-Mengerink et al 2013, Huffnagel et al 2013]; in a recent study 11 of 12 affected individuals survived to adulthood [Duker et al 2020].
Skeletal. Most individuals with mild RCDP1 have limited joint mobility due to flexion contractures of the elbows, knees, and hips. While the radiographic finding of CDP (chondrodysplasia punctata) is commonly noted at the time of RCDP1 diagnosis, rhizomelic limb shortening is uncommon in this group. Deformities in hip joints including coxa vara and small femoral heads have been reported (see Figure 6) [Barth et al 1996, Bams-Mengerink et al 2013]. Many of these individuals required orthopedic surgeries over time to improve mobility and activities of daily living [Barth et al 1996].
Cataract. Most individuals with mild RCDP1 have bilateral cataracts diagnosed in the first two years of life.
Growth of individuals with mild RCDP1 can be within normal ranges at birth. Postnatal growth rates are also better than those in classic RCDP1. Growth curves based on four individuals with nonclassic RCDP1 have been published [Duker et al 2017].
Intellect. Most individuals with mild RCDP1 have had developmental delays and learning disabilities. However, they were able to achieve gross and fine motor skills never achieved in individuals with classic RCDP1. All individuals with mild RCDP1 were able to walk and most can communicate verbally; all required some degree of special education. Brain MRI was normal in all three individuals with nonclassic RCDP1 reported by Bams-Mengerink et al [2006].
Seizures. In one cohort, three of four individuals with mild RCDP1 developed seizures in late childhood (age range 7-21 years). The type of seizures more commonly seen in this group were absence and tonic-clonic seizures [Bams-Mengerink et al 2013].
Congenital heart disease. In six individuals with mild RCDP1, cardiac defects including atrial septal defects were reported in two individuals, one of whom also had first-degree heart block. Two individuals developed mitral valve prolapse, possibly indicating degenerative cardiac changes [Huffnagel et al 2013].
Behavior disorders, usually identified at school age in the limited number of individuals described with mild RCDP1, included attention-deficit/hyperactivity disorder and autism spectrum disorder (ASD) [Bams-Mengerink et al 2013, Yu et al 2013].
In one family, two sibs with the PEX7 variant p.Ser25Phe had congenital cataract. One sib had ASD, intellectual disability, and epilepsy; the other had normal intellect with attention-deficit disorder. Elevated blood phytanic acid was observed on an unrestricted diet.
In another family, two sibs had the PEX7 variant p.Trp75Cys and ASD, intellectual disability, epilepsy, and cataracts.
Retinitis pigmentosa and peripheral neuropathy. An individual with the PEX7 variant c.-45C>T had developmental delays and poor growth in childhood; retinitis pigmentosa and peripheral neuropathy developed in adolescence [Braverman et al 2002]. Cataracts were not reported.
Genotype-Phenotype Correlations
Correlations between the predicted severity of PEX7 pathogenic variants and RCDP1 phenotype include the following:
- All individuals homozygous for the p.Leu292Ter pathogenic variant studied to date have had classic RCDP1.
- In individuals who are compound heterozygotes for p.Leu292Ter and another pathogenic variant, the effect of the other allele is important in determining the phenotype. Several PEX7 variants that are associated with a milder RCDP1 phenotype or adult Refsum disease have been identified. It is predicted that these encode either residual amounts of a normal PEX7 protein or a defective protein with residual function [Braverman et al 2002, Motley et al 2002, van den Brink et al 2003].
Nomenclature
RCDP1 is one of two groups of peroxisome biogenesis disorders (PBD); the other group is Zellweger spectrum disorder.
Although individuals with RCDP1 have a perturbation in matrix protein import consistent with a peroxisomal assembly defect, they have a biochemical, cellular, and clinical phenotype distinct from Zellweger spectrum disorder.
Prevalence
The prevalence of RCDP1 is estimated to be lower than 1:100,000.
The high frequency of the p.Leu292Ter variant is secondary to a founder effect in individuals of northern European descent [Braverman et al 2000]. The p.Leu292Ter allele was recently identified in high frequency in a genetically isolated community in the Netherlands where the carrier frequency among tested individuals was around 6% [Mathijssen et al 2015]. See Table 6.
Genetically Related (Allelic) Disorders
Adult Refsum disease. Biallelic PEX7 pathogenic variants can result in adult Refsum disease. In PEX7-deficient individuals described with this disorder, plasmalogen biosynthesis was normal or near normal; however, phytanic acid oxidation in skin fibroblast assays was severely reduced [Braverman et al 2002, van den Brink et al 2003].
An individual with the PEX7 variant (reference sequence NM_000288.3:c.40A>C;p.Thr14Pro) had congenital cataracts and later developed adult Refsum disease [van den Brink et al 2003].
Additional PEX7 variants associated with adult Refsum disease are c.12_18dup [van den Brink et al 2003] and c.340-10A>G (IVS3-10A>G) [Braverman et al 2002] (reference sequence NM_000288.3).
Thus, it is likely that a continuum of PEX7 phenotypes will emerge, reflecting a PEX7 disorder spectrum that includes classic (severe) RCDP1, nonclassic (mild) RCDP1, and adult Refsum disease-like phenotypes.
Differential Diagnosis
Table 2.
Genes of Interest in the Differential Diagnosis of Rhizomelic Chondrodysplasia Punctata Type 1 (RCDP1)
Other disorders to consider in the differential diagnosis of RCDP1
- Chondrodysplasia punctata, tibia-metacarpal type (OMIM 118651) and humero-metacarpal type [Fryburg & Kelly 1996] are inherited in an autosomal dominant manner. The gene(s) in which pathogenic variants are causative are unknown. Affected individuals have short metacarpals with shortening of various long bones. No cataracts or skin changes are present.
- Warfarin embryopathy and other fetal vitamin K deficiencies (including vitamin K epoxide reductase deficiency; see Table 4) show CDP, but are distinguished clinically from RCDP1 by nasomaxillary hypoplasia, brachytelephalangy, and absence of rhizomelia and cataracts. With the exception that males and females are affected, this phenotype is similar to X-linked chondrodysplasia punctata 1 (CDPX1).
- Maternal systemic lupus erythematosus (SLE) (OMIM 152700) and other maternal autoimmune diseases can cause CDP in the offspring, but are distinguished from RCDP1 by nasomaxillary hypoplasia, brachytelephalangy and absence of rhizomelia and cataracts. With the exception that males and females are affected, this phenotype is similar to CDPX1.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with rhizomelic chondrodysplasia punctata type I (RCDP1), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Rhizomelic Chondrodysplasia Punctata Type 1
Treatment of Manifestations
In classic (severe) RCDP1, management is supportive because of the multiple handicaps present at birth and the poor outcome.
In children with nonclassic (mild) RCDP1, orthopedic surgeries have been performed to maintain gait and joint mobility. Individual education plans have enabled these children to maximize benefit from the school system.
Table 4.
Treatment of Manifestations in Individuals with Rhizomelic Chondrodysplasia Punctata Type 1
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states and provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center-based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Social/Behavioral Concerns
Children with nonclassic (mild) RCDP1 may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
Table 5.
Recommended Surveillance for Individuals with Rhizomelic Chondrodysplasia Punctata Type 1
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Studies in rodents suggest that plasmalogen precursor supplementation using alkylglycerol sources can increase plasmalogen concentrations in somatic tissues but not in the nervous system [Braverman et al 2010, Brites et al 2011, Malheiro et al 2019].
Anecdotal reports of alkylglycerol supplementation in a few individuals with classic RCDP1 have not indicated dramatic clinical benefit; however, alkylglycerol supplementation in humans has not yet been studied in a systematic fashion.
Nonsense suppressor drugs were unable to recover protein production in individuals with the common p.Leu292Ter variant [Dranchak et al 2011].
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Rhizomelic chondrodysplasia punctata type 1 (RCDP1) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one PEX7 pathogenic variant).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for a PEX7 pathogenic variant and to allow reliable recurrence risk assessment. (De novo variants are known occur at a low but appreciable rate in autosomal recessive disorders [Jónsson et al 2017].)
- Heterozygotes are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for a PEX7 pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting both pathogenic variants and being affected, a 50% chance of inheriting one pathogenic variant and being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. To date, individuals with RCDP1 are not known to reproduce.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a PEX7 pathogenic variant.
Carrier Detection
Molecular genetic carrier testing for at-risk relatives requires prior identification of the PEX7 pathogenic variants in the family.
Note: Carriers cannot be identified by biochemical methods.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are carriers or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the PEX7 pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Biochemical testing. Prenatal testing for pregnancies at 25% risk for RCDP1 is also possible by assay of plasmalogen biosynthesis in cultured chorionic villi obtained by CVS (usually performed at ~10-12 weeks' gestation) or in cultured amniocytes obtained by amniocentesis (usually performed at ~15-18 weeks' gestation).
Ultrasound examination. Rhizomelia and punctate calcifications have been noted on ultrasound examination as early as 18 to 19 weeks [Landino et al 2017, Krakow et al 2003, Zwijnenburg et al 2010]. Others have reported these findings along with bilateral cataracts at 32 weeks, and epiphyseal stippling shown four weeks later [Başbuğ et al 2005]. Homozygosity of the p.Leu292Ter variant in PEX7 was verified in the case reported by Zwijnenburg et al [2010].
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- RhizoKids
- Little People of AmericaPhone: 888-LPA-2001; 714-368-3689Fax: 707-721-1896Email: info@lpaonline.org
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Rhizomelic Chondrodysplasia Punctata Type 1: Genes and Databases
Table B.
OMIM Entries for Rhizomelic Chondrodysplasia Punctata Type 1 (View All in OMIM)
Molecular Pathogenesis
RDCP1 is a disorder of peroxisome biogenesis, caused by defects in PEX7. PEX7 protein is a receptor for peroxisomal matrix enzymes that contain a PTS2 targeting sequence. The enzymes:
- AGPS (required for plasmalogen synthesis)
- PHYH (required for phytanic acid oxidation)
- ACAA1 (involved in fatty acid B oxidation, but not required)
PEX7 binds these enzymes to the PEX5 receptor (which binds PTS1-targeted enzymes) in the cytosol to carry its cargo to the peroxisome membrane, where the enzymes are translocated inside. Subsequently, the import complex is disassembled, and the PEX7 receptor and other proteins of the complex are recycled for another round of import. This import process, along with the formation of new peroxisomes and division of existing ones, is termed peroxisome biogenesis and requires the coordinated action of multiple PEX proteins. Defects in any one of 13 of these cause the peroxisome biogenesis disorder Zellweger spectrum. See Kunze et al [2011], Waterham & Ebberink [2012], and Waterham et al [2016] for details of PEX genes, peroxisome biogenesis, and disorders of peroxisome deficiency.
Mechanism of disease causation. Pathogenic variants in PEX7 impair the import of PTS2-targeted peroxisome matrix enzymes resulting in deficient activity of these enzymes; however, other peroxisomal functions remain intact, such as import of PTS1-targeted proteins by PEX5 and peroxisome membrane assembly. In vitro assays in fibroblasts show that the PTS2-targeted peroxisomal matrix enzymes remain cytosolic and are degraded in individuals with RCDP1. There is no evidence to support gain-of-function variants as a cause of RCDP1.
Variants associated with milder RCDP1 phenotype or adult Refsum disease are either:
- Missense variants located on the surface of the PEX7 protein and, thus, unlikely to disrupt its structural integrity (p.Ser25Phe, p.His285Arg, p.Thr14Pro); OR
- "Leaky" alleles, potentially able to generate residual amounts of normal PEX7 protein (c.-45C>T, c.340-10A>G) or to reinitiate translation in-frame (p.His18ArgfsTer35) or located at a downstream methionine residue (p.Gly7ValfsTer51) (reference sequences NM_000288.3, NP_000279.1) [Braverman et al 2002, Motley et al 2002, van den Brink et al 2003].
PEX7-specific laboratory technical considerations. Technical considerations in sequencing PEX7 include reviewing the 5'UTR and intron 3 for the reported variants in Table 6.
Notable PEX7 variants. Exon and multiexon deletions in PEX7 have been identified [N Braverman, unpublished].
Table 6.
Notable PEX7 Pathogenic Variants
Chapter Notes
Author History
Michael B Bober, PhD, MD (2020-present)
Nancy E Braverman, MS, MD (2001-present)
Angela Duker, MS, CGC (2020-present)
Wedad Fallatah, MD, MAS (2020-present)
Ann B Moser, BA; Kennedy Krieger Institute (2001-2020)
Steven K Steinberg, PhD (2001-present)
Revision History
- 30 January 2020 (bp) Comprehensive update posted live
- 13 September 2012 (me) Comprehensive update posted live
- 2 March 2010 (me) Comprehensive update posted live
- 18 July 2006 (me) Comprehensive update posted live
- 7 February 2005 (cd) Revision: test availability
- 26 February 2004 (cd) Revision: test availability
- 13 February 2004 (me) Comprehensive update posted live
- 16 November 2001 (me) Review posted live
- 10 June 2001 (nb) Original submission
References
Literature Cited
- Abousamra O, Kandula V, Duker AL, Rogers KJ, Bober MB, Mackenzie WG. Cervical spine deformities in children with rhizomelic chondrodysplasia punctata. J Pediatr Orthop. 2019;39:e680–e686. [PubMed: 31503224]
- Alkan A, Kutlu R, Yakinci C, Sigirci A, Aslan M, Sarac K. Delayed myelination in a rhizomelic chondrodysplasia punctata case: MR spectroscopy findings. Magn Reson Imaging. 2003;21:77–80. [PubMed: 12620550]
- Bams-Mengerink AM, Koelman JH, Waterham H, Barth PG, Poll-The BT. The neurology of rhizomelic chondrodysplasia punctata. Orphanet J Rare Dis. 2013;8:174. [PMC free article: PMC4228450] [PubMed: 24172221]
- Bams-Mengerink AM, Majoie CBLM, Duran M, Wanders RJA, Van Hove J, Scheurer CD, Barth PG, Poll-The BT. MRI of the brain and certical spinal cord in rhizomelic chondrodysplasia punctata. Neurology. 2006;66:798–803. [PubMed: 16567694]
- Barth PG, Wanders RJ, Schutgens RB, Staalman CR. Variant rhizomelic chondrodysplasia punctata (RCDP) with normal plasma phytanic acid: clinico-biochemical delineation of a subtype and complementation studies. Am J Med Genet. 1996;62:164–8. [PubMed: 8882397]
- Başbuğ M, Serin IS, Ozçelik B, Guneş T, Akçakuş M, Tayyar M. Prenatal ultrasonographic diagnosis of rhizomelic chondrodysplasia punctata by detection of rhizomelic shortening and bilateral cataracts. Fetal Diagn Ther. 2005;20:171–4. [PubMed: 15824492]
- Braverman N, Chen L, Lin P, Obie C, Steel G, Douglas P, Chakraborty PK, Clarke JT, Boneh A, Moser A, Moser H, Valle D. Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype. Hum Mutat. 2002;20:284–97. [PubMed: 12325024]
- Braverman N, Steel G, Lin P, Moser A, Moser H, Valle D. PEX7 gene structure, alternative transcripts, and evidence for a founder haplotype for the frequent RCDP allele, L292ter. Genomics. 2000;63:181–92. [PubMed: 10673331]
- Braverman N, Zhang R, Chen L, Nimmo G, Scheper S, Tran T, Chaudhury R, Moser A, Steinberg S. A. Pex7 hypomorphic mouse model for plasmalogen deficiency affecting the lens and skeleton. Mol Genet Metab. 2010;99:408–16. [PMC free article: PMC2839039] [PubMed: 20060764]
- Braverman NE, Moser AB. Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta. 2012;1822:1442–52. [PubMed: 22627108]
- Brites P, Ferreira AS, da Silva TF, Sousa VF, Malheiro AR, Duran M, Waterham HR, Baes M, Wanders RJ. Alkyl-glycerol rescues plasmalogen levels and pathology of ether-phospholipid deficient mice. PLoS One. 2011;6:e28539. [PMC free article: PMC3232224] [PubMed: 22163031]
- Dranchak PK, Di Pietro E, Snowden A, Oesch N, Braverman NE, Steinberg SJ, Hacia JG. Nonsense suppressor therapies rescue peroxisome lipid metabolism and assembly in cells from patients with specific PEX gene mutations. J Cell Biochem. 2011;112:1250–8. [PMC free article: PMC3136445] [PubMed: 21465523]
- Duker AL, Eldridge G, Braverman NE, Bober MB. Congenital heart defects common in rhizomelic chondrodysplasia punctata. Am J Med Genet A. 2016;170A:270–2. [PubMed: 26408048]
- Duker AL, Niiler T, Eldridge G, Brereton NH, Braverman NE, Bober MB. Growth charts for individuals with rhizomelic chondrodysplasia punctata. Am J Med Genet A. 2017;173:108–13. [PubMed: 27616591]
- Duker AL, Niiler T, Schouten M, Poll-The BT, Braverman NE, Bober MB. Rhizomelic chondrodysplasia punctata morbidity and mortality, an update. Am J Med Genet A. 2020;182:579–83. [PubMed: 31769196]
- Fryburg JS, Kelly TE. Chondrodysplasia punctata, humero-metacarpal type: a second case. Am J Med Genet. 1996;64:493–6. [PubMed: 8862628]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Huffnagel IC, Clur SA, Bams-Mengerink AM, Blom NA, Wanders RJ, Waterham HR, Poll-The BT. Rhizomelic chondrodysplasia punctata and cardiac pathology. J Med Genet. 2013;50:419–24. [PubMed: 23572185]
- Irving MD, Chitty LS, Mansour S, Hall CM. Chondrodysplasia punctata: a clinical diagnostic and radiological review. Clin Dysmorphol. 2008;17:229–41. [PubMed: 18978650]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Khanna AJ, Braverman NE, Valle D, Sponseller PD. Cervical stenosis secondary to rhizomelic chondrodysplasia punctata. Am J Med Genet. 2001;99:63–6. [PubMed: 11170096]
- Krakow D, Williams J 3rd, Poehl M, Rimoin DL, Platt LD. Use of three-dimensional ultrasound imaging in the diagnosis of prenatal-onset skeletal dysplasias. Ultrasound Obstet Gynecol. 2003;21:467–72. [PubMed: 12768559]
- Kunze M, Neuberger G, Maurer-Stroh S, Ma J, Eck T, Braverman N, Schmid JA, Eisenhaber F, Berger J. Structural requirements for interaction of peroxisomal targeting signal 2 and its receptor PEX7. J Biol Chem. 2011;286:45048–62. [PMC free article: PMC3247985] [PubMed: 22057399]
- Landino J, Jnah AJ, Newberry DM, Iben SC. Neonatal rhizomelic chondrodysplasia punctata type 1: weaving evidence into clinical practice. J Perinat Neonatal Nurs. 2017;31:350–7. [PubMed: 29068853]
- Malheiro AR, Correia B, Ferreira da Silva T, Bessa-Neto D, Van Veldhoven PP, Brites P. Leukodystrophy caused by plasmalogen deficiency rescued by glyceryl 1-myristyl ether treatment. Brain Pathol. 2019;29:622–39. [PMC free article: PMC8028673] [PubMed: 30667116]
- Mathijssen IB, Henneman L, van Eeten-Nijman JM, Lakeman P, Ottenheim CP, Redeker EJ, Ottenhof W, Meijers-Heijboer H, van Maarle MC. Targeted carrier screening for four recessive disorders: high detection rate within a founde population. Eur J Med Genet. 2015;58:123–8. [PubMed: 25641760]
- Motley AM, Brites P, Gerez L, Hogenhout E, Haasjes J, Benne R, Tabak HF, Wanders RJ, Waterham HR. Mutational spectrum in the PEX7 gene and functional analysis of mutant alleles in 78 patients with rhizomelic chondrodysplasia punctata type 1. Am J Hum Genet. 2002;70:612–24. [PMC free article: PMC384941] [PubMed: 11781871]
- Oswald G, Lawson C, Raymond G, Golden WC, Braverman N. Rhizomelic chondrodysplasia punctata type 1 and fulminant neonatal respiratory failure, a case report and discussion of pathophysiology. Am J Med Genet A. 2011;155A:3160–3. [PubMed: 22052861]
- Powers JM, Kenjarski TP, Moser AB, Moser HW. Cerebellar atrophy in chronic rhizomelic chondrodysplasia punctata: a potential role for phytanic acid and calcium in the death of its Purkinje cells. Acta Neuropathol (Berl). 1999;98:129–34. [PubMed: 10442551]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Smeitink JA, Beemer FA, Espeel M, Donckerwolcke RA, Jakobs C, Wanders RJ, Schutgens RB, Roels F, Duran M, Dorland L, Berger R, Poll-The BT. Bone dysplasia associated with phytanic acid accumulation and deficient plasmalogen synthesis: a peroxisomal entity amenable to plasmapheresis. J Inher Metab Dis. 1992;15:377–80. [PubMed: 1405474]
- van den Brink DM, Brites P, Haasjes J, Wierzbicki AS, Mitchell J, Lambert-Hamill M, de Belleroche J, Jansen GA, Waterham HR, Wanders RJ. Identification of PEX7 as the second gene involved in Refsum disease. Am J Hum Genet. 2003;72:471–7. [PMC free article: PMC379239] [PubMed: 12522768]
- Waterham HR, Ebberink MS. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim Biophys Acta. 2012;1822:1430–41. [PubMed: 22871920]
- Waterham HR, Ferdinandusse S, Wanders RJ. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta. 2016;1863:922–33. [PubMed: 26611709]
- White AL, Modaff P, Holland-Morris F, Pauli RM. Natural history of rhizomelic chondrodysplasia punctata. Am J Med Genet. 2003;118A:332–42. [PubMed: 12687664]
- Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, Schmitz-Abe K, Harmin DA, Adli M, Malik AN, D'Gama AM, Lim ET, Sanders SJ, Mochida GH, Partlow JN, Sunu CM, Felie JM, Rodriguez J, Nasir RH, Ware J, Joseph RM, Hill RS, Kwan BY, Al-Saffar M, Mukaddes NM, Hashmi A, Balkhy S, Gascon GG, Hisama FM, LeClair E, Poduri A, Oner O, Al-Saad S, Al-Awadi SA, Bastaki L, Ben-Omran T, Teebi AS, Al-Gazali L, Eapen V, Stevens CR, Rappaport L, Gabriel SB, Markianos K, State MW, Greenberg ME, Taniguchi H, Braverman NE, Morrow EM, Walsh CA. Using whole exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–73. [PMC free article: PMC3694430] [PubMed: 23352163]
- Zwijnenburg PJ, Deurloo KL, Waterham HR, Meijers-Heijboer EJ, van Vugt JM, Tan-Sindhunata MB. Second trimester prenatal diagnosis of rhizomelic chondrodysplasia punctata type 1 on ultrasound findings. Prenat Diagn. 2010;30:162–4. [PubMed: 20014169]
Publication Details
Author Information and Affiliations
McGill University;
Montreal Children's Hospital Research Institute
Montreal, Quebec, Canada
McGill University
Montreal, Quebec, Canada
Nemours / Alfred I duPont Hospital for Children
Wilmington, Delaware
Nemours / Alfred I duPont Hospital for Children
Wilmington, Delaware
Publication History
Initial Posting: November 16, 2001; Last Update: January 30, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Braverman NE, Steinberg SJ, Fallatah W, et al. Rhizomelic Chondrodysplasia Punctata Type 1. 2001 Nov 16 [Updated 2020 Jan 30]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.