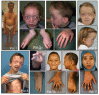
Clinical Description
To date, 44 individuals have been identified with a pathogenic variant in SKI [Carmignac et al 2012, Doyle et al 2012, Au et al 2014, Schepers et al 2015, Saito et al 2017, O'Dougherty et al 2019, Zhang et al 2019]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Select Features of Shprintzen-Goldberg Syndrome
View in own window
| Feature | # of Persons w/Feature /
# Evaluated for Feature |
|---|
| Developmental delay / intellectual disability | 41/44 |
| Hypotonia | 16/19 |
| Craniosynostosis 1 | 31/41 |
| Dolichocephaly/scaphocephaly | 36/39 |
| Hypertelorism | 42/43 |
| Downslanting palpebral fissures | 38/41 |
| Ocular proptosis | 34/42 |
| Malar flattening | 24/24 |
| High narrow palate | 23/23 |
| Micrognathia | 36/40 |
| Low-set, posteriorly rotated ears | 23/24 |
| Arachnodactyly | 43/44 |
| Camptodactyly | 24/38 |
| Pectus deformity | 32/40 |
| Scoliosis | 29/39 |
| Joint hypermobility | 18/22 |
| Joint contractures | 32/36 |
| Foot malposition / talipes equinovarus / club foot / pes cavus | 23/31 |
| C1-C2 spine malformation | 7/10 |
| Aortic root dilatation | 14/41 |
| Mitral valve prolapse / valvular anomalies | 12/38 |
| Abdominal hernias | 14/23 |
| Minimal subcutaneous fat / marfanoid habitus | 9/20 |
- 1.
Typically coronal, sagittal, or lambdoid sutures
Neurodevelopment. Motor and cognitive milestones are delayed and intellectual disability is usually mild to moderate. To date, three individuals with Shprintzen-Goldberg syndrome (SGS) and a confirmed SKI pathogenic variant were reported to have normal intelligence [Doyle et al 2012, Schepers et al 2015, Zhang et al 2019].
Craniosynostosis usually involves the coronal, sagittal, or lambdoid sutures. The sutures are fused at birth and craniosynostosis can usually be suspected from the abnormal skull shape. A single suture or multiple sutures may be involved [Au et al 2014]. Information on individuals with SGS who underwent surgery for craniosynostosis in unavailable.
Characteristic craniofacial features include hypertelorism, downslanting palpebral fissures, ocular proptosis, high narrow palate, micrognathia, and low-set posteriorly rotated ears. Cleft palate was reported in three of 17 individuals with SGS [Doyle et al 2012, Schepers et al 2015, O'Dougherty et al 2019]. Broad/bifid uvula has been reported in two of 12 individuals [Doyle et al 2012, O'Dougherty et al 2019].
Musculoskeletal. Arachnodactyly, camptodactyly, pectus deformities, and clubfeet are common features and are present at birth. Joint contractures are often present at birth or in the neonatal period, with the ankle joint most frequently affected [Au et al 2014, Saito et al 2017]. Joint hypermobility also occurs in individuals with SGS and is typically present from birth. Joint dislocation and instability reported by O'Dougherty et al [2019] included involvement of the left patella, thumb, and foot and C1-C2 instability. Au et al [2014] reported subluxing patellae. Pes planus becomes evident later in childhood; one or both feet may be affected. Scoliosis may be severe [Au et al 2014].
Cardiovascular. Aortic root dilatation was present in three of 18 affected individuals reported by Carmignac et al [2012]. In the report of Doyle et al [2012], however, eight of ten individuals with SGS and confirmed pathogenic variants in SKI had aortic root dilatation with or without mitral valve prolapse/incompetence. Surgery at age 16 years for aortic dilatation (aortic root dilatation with z score = 7.014) was reported in one individual with molecularly confirmed SGS [Carmignac et al 2012]. This individual also had vertebrobasilar and internal carotid tortuosity and a dilated pulmonary artery root. Among the affected individuals with molecularly confirmed SGS reported by Doyle et al [2012] one had arterial tortuosity and two had splenic artery aneurysm – one with spontaneous rupture.
Ocular. Myopia was reported in ten of 19 individuals [Carmignac et al 2012, Au et al 2014, O'Dougherty et al 2019]. Ectopia lentis has not been reported in the 16 individuals with SGS who were evaluated for this finding [Doyle et al 2012, Schepers et al 2015].
Minimal subcutaneous fat and/or marfanoid habitus was reported in nine of 20 individuals [Carmignac et al 2012, Au et al 2014].
Abdominal wall defects were reported in 14 of 23 individuals [Carmignac et al 2012, Au et al 2014, Schepers et al 2015, Saito et al 2017, O'Dougherty et al 2019].
Other
Nomenclature
Goldberg-Shprintzen syndrome and Shprintzen-Goldberg omphalocele syndrome are separate syndromes, not related to SGS.
Other names that have been used to refer to SGS:
Craniosynostosis with arachnodactyly and abdominal hernias
Marfanoid-craniosynostosis syndrome
Shprintzen-Goldberg craniosynostosis syndrome
Shprintzen-Goldberg marfanoid syndrome
The term Furlong syndrome has been used to describe one individual with craniosynostosis, features of SGS, normal intelligence, and aortic enlargement. Adès et al [2006] reported on two individuals with a phenotype similar to Furlong syndrome. They had the same pathogenic missense variant in TGFBR1, making a diagnosis of Loeys-Dietz syndrome type 1 most likely [B Loeys, personal communication]. In the absence of analysis for pathogenic variants in the original individual described as having Furlong syndrome, the existence of this as a separate entity remains unclear.