Summary
Clinical characteristics.
Hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC), a neurodevelopmental and neurodegenerative disorder, is characterized by severe progressive sensorimotor neuropathy with resulting hypotonia, areflexia, and amyotrophy, and by variable degrees of dysgenesis of the corpus callosum. Mild-to-severe intellectual disability and "psychotic episodes" during adolescence are observed. Sensory modalities are moderately to severely affected beginning in infancy. The average age of onset of walking is 3.8 years; the average age of loss of walking is 13.8 years; the average age of death is 33 years.
Diagnosis/testing.
The diagnosis of HMSN/ACC is established in a proband with suggestive findings and biallelic pathogenic variants in SLC12A6 identified by molecular genetic testing.
Management.
Treatment of manifestations: Walking aids such as canes or walkers are required. As the disease progresses, orthoses for upper and lower limbs and physiotherapy are needed to prevent contractures. Early developmental/educational intervention addresses cognitive delays. Depending on severity, individuals with HMSN/ACC usually require corrective surgery for scoliosis. Neuroleptics may be used to treat psychiatric manifestations, usually during adolescence.
Surveillance: Monitor in the early teens for scoliosis and in the late teens for psychiatric manifestations.
Genetic counseling.
HMSN/ACC is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Heterozygotes (carriers) are asymptomatic. Once the SLC12A6 pathogenic variants have been identified in an affected family member, carrier testing for at-risk family members and prenatal and preimplantation genetic testing are possible.
Diagnosis
Consensus diagnostic criteria for hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC) have not been established.
Suggestive Findings
Hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC) should be suspected in individuals with the following clinical, electrophysiologic, and neuroimaging findings, and family history [Dupré et al 2003].
Clinical findings
- Severe progressive sensorimotor neuropathy with areflexia
- Developmental delay / intellectual disability ranging from mild to severe
Electrophysiology
- Sensory nerve action potentials cannot be recorded at the median, ulnar, or sural nerves even in children in their first year of life.
- Compound motor action potentials usually show diminished amplitudes.
- Nerve conduction velocities for the median, ulnar, and tibial nerves are variable.
- Needle electromyography may show mild signs of active denervation (e.g., fibrillation potentials).
Neuroimaging
- Mild cortical or cerebellar atrophy at a later age
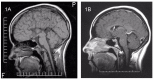
Figure 1.
Sagittal T1-weighted MRI A. Complete agenesis of the corpus callosum

Figure 2.
Axial T1-weighted MRI A. Agenesis of the corpus callosum with parallelism of the ventricles
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC) is established in a proband with suggestive findings and biallelic pathogenic (or likely pathogenic) variants in SLC12A6 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic SLC12A6 variants of uncertain significance (or identification of one known SLC12A6 pathogenic variant and one SLC12A6 variant of uncertain significance) does not establish or rule out a diagnosis of this disorder.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing or multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of HMSN/ACC has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of SLC12A6 is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
Note: In individuals of French Canadian origin who have the typical phenotype, targeted analysis for the c.2436+1delG pathogenic variant can be performed first, followed by sequence analysis if only one or no variant is identified.
An agenesis of the corpus callosum panel, an intellectual disability panel, or a comprehensive neuropathy panel (multigene panels that include SLC12A6 and other genes of interest; see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Hereditary Motor and Sensory Neuropathy with Agenesis of the Corpus Callosum
Clinical Characteristics
Clinical Description
Hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC) is both a neurodevelopmental disorder (with variable degrees of dysgenesis of the corpus callosum and mild-to-severe intellectual disability) and a neurodegenerative disorder (severe progressive sensorimotor neuropathy).
The neurologic findings of HMSN/ACC in 64 individuals (ages 2 to 34 years) in the French Canadian population reported by Mathieu et al [1990] are summarized in Table 2, with additional information from Larbrisseau et al [1984], Salin-Cantegrel et al [2007], and Auer et al [2016].
Table 2.
Hereditary Motor and Sensory Neuropathy with Agenesis of the Corpus Callosum: Select Features
Progressive motor and sensory neuropathy
- Reflexes invariably absent from infancy
- Hypotonia invariably present in the first year of life
- Progressive distal and proximal symmetric limb weakness
- Muscle atrophy
- Diffuse limb tremor (probably secondary to polyneuropathy)
- Contractures
- Loss of sensation to touch and pinprick in a glove and stocking distribution; easier to evaluate in older children
The average age of onset for sitting alone is 2.1 years, for standing is more than two years, for walking is 3.8 years, and average age of loss of ability to walk is 13.8 years.
Cognitive function. The Taft clinical classification uses an IQ test to rank individuals in four categories: normal intelligence (IQ >75), mild intellectual disability (IQ = 50-75), moderate intellectual disability (IQ = 25-50), and severe intellectual disability (IQ <25). Individuals with mild intellectual disability can achieve five to six years of elementary school and live independently. Individuals with moderate intellectual disability can help with activities of daily living but require supervision on a daily basis. Individuals with severe intellectual disability require assistance for daily living and supervision but may be able to take care of some routine daily needs. The range of intelligence of individuals with HSMN/ACC is normal IQ to severe intellectual disability.
Psychotic episodes. Mathieu et al [1990] reported that after age 15 years, 39% (25/64) developed "psychotic episodes" characterized by paranoid delusions, depressive states, visual hallucinations, auditory hallucinations, or "autistic-like" features.
Life expectancy. Average age of death is 33 years and is usually related to respiratory insufficiency [Larbrisseau & Sarnat 2017].
Other
- Lumbar puncture usually reveals mild elevation of CSF proteins [Dupré et al 2003].
- Sural nerve biopsy shows an almost total lack of large myelinated fibers, signs of axonal loss (ovoids of Wallerian degeneration), and some enlarged axons that on electron microscopy show decreased density of neurofilaments. Isolated fibers may have disproportionately thin myelin sheaths, suggesting that the axoplasm is swollen. Electron microscopy may show decreased packing density of neurofilaments, without signs of their degradation [Dupré et al 2003, Auer et al 2016].Note: Sural nerve biopsy is unnecessary to confirm the diagnosis, now that molecular genetic testing is possible.
- EEG may be normal or with epileptiform abnormalities [Dupré et al 2003, Salin-Cantegrel et al 2007].
- Muscle biopsy shows nonspecific signs of chronic denervation atrophy [Dupré et al 2003, Auer et al 2016].
Autopsy examination. The hallmark is swollen axons in cranial nerve samples (especially cranial nerves 3 and 7), as well as in the dorsal and ventral nerve roots. Swollen axons can also be scattered in the white matter. The brain shows either no agenesis of the corpus callosum (ACC), partial ACC, or complete ACC with preservation of Probst bundle [Dupré et al 2003, Auer et al 2016].
Genotype-Phenotype Correlations
The data from affected individuals are insufficient to establish genotype-phenotype correlations.
Nomenclature
HMSN/ACC may also be referred to as Charlevoix disease.
Prevalence
In the French Canadian population of the Saguenay and Lac-St-Jean regions of Quebec, Canada, the overall incidence of HMSN/ACC is 1:2,117 live births; the carrier rate is 1:23 inhabitants due to the c.2436+1delG (also known as c.2436delG) founder variant. Otherwise, HMSN/ACC is extremely rare worldwide. Three pairs of sibs (of Italian, Mexican, and Turkish origin) have been reported to have molecularly confirmed HMSN/ACC.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with mutation of SLC12A6.
Differential Diagnosis
Table 3.
Autosomal Recessive Neurodegenerative Disorders in the Differential Diagnosis of Hereditary Motor and Sensory Neuropathy with Agenesis of the Corpus Callosum
Management
Consensus clinical management recommendations for hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC) have not been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC), the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Hereditary Motor and Sensory Neuropathy with Agenesis of the Corpus Callosum
Treatment of Manifestations
Care is best provided by a multidisciplinary team that comprises a pediatrician or pediatric neurologist, developmental pediatrician, psychiatrist, orthopedist, physiotherapist, and occupational therapist.
Table 5.
Treatment of Manifestations in Individuals with Hereditary Motor and Sensory Neuropathy with Agenesis of the Corpus Callosum
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Surveillance
Table 6.
Recommended Surveillance for Individuals with Hereditary Motor and Sensory Neuropathy with Agenesis of the Corpus Callosum
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., presumed to be carriers of one SLC12A6 pathogenic variant based on family history).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for an SLC12A6 pathogenic variant and to allow reliable recurrence risk assessment. (De novo variants are known to occur at a low but appreciable rate in autosomal recessive disorders [Jónsson et al 2017].)
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for an SLC12A6 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. Due to the severity of the disorder, individuals with HMSN/ACC do not reproduce.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an SLC12A6 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the SLC12A6 pathogenic variants in the family.
See Related Genetic Counseling Issues, Population screening for information about carrier testing in individuals who do not have a family history of HMSN/ACC.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of genetic status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are carriers or are at risk of being carriers.
Population screening. Individuals with a genealogic link to regions with high HMSN/ACC carrier rates may choose to have carrier testing for the c.2436+1delG French Canadian HMSN/ACC founder variant (see Prevalence and Table 7).
Prenatal Testing and Preimplantation Genetic Testing
Once the SLC12A6 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- National Organization for Disorders of the Corpus CallosumEmail: info@nodcc.org
- Hereditary Neuropathy FoundationPhone: 855-435-7268 (toll-free); 212-722-8396Fax: 917-591-2758Email: info@hnf-cure.org
- Muscular Dystrophy CanadaCanadaPhone: 800-567-2873Email: info@muscle.ca
- National Institute of Neurological Disorders and Stroke (NINDS)PO Box 5801Bethesda MD 20824Phone: 800-352-9424 (toll-free); 301-496-5751; 301-468-5981 (TTY)
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Hereditary Motor and Sensory Neuropathy with Agenesis of the Corpus Callosum: Genes and Databases
Table B.
OMIM Entries for Hereditary Motor and Sensory Neuropathy with Agenesis of the Corpus Callosum (View All in OMIM)
Molecular Pathogenesis
SLC12A6 encodes solute carrier family 12 member 6 (KCC3) whose protein structure includes:
- 12 putative membrane-spanning helices with large NH2 and COOH termini;
- A large extracellular loop between transmembrane domains 7 and 8 with five potential sites for N-linked glycosylation;
- Two consensus cAMP-dependant protein kinase phosphorylation sites;
- Four consensus protein kinase C phosphorylation sites in the COOH terminus.
KCC3, a cell cotransporter of the peripheral and central nervous system throughout neurologic development and adulthood, is necessary for the transport of chloride and potassium for the regulation of axonal cell volume [Delpire & Kahle 2017, Flores et al 2019]. Absence of KCC3 alters maintenance of homeostasis of the membrane threshold, leading to neuronal degeneration [Flores et al 2019].
KCC3 is also involved in the precise propagation of the paranodal action potential [Sun et al 2016]. This is consistent with brain autopsies of individuals with HMSN/ACC in whom regenerative clusters in the peripheral nervous system indicate ongoing degeneration [Auer et al 2016].
Although both loss of function and gain of function of KCC3 can cause peripheral neuropathy, gain of KCC3 function causes peripheral neuropathy without abnormalities of the corpus callosum or cognitive abilities [Flores et al 2019].
Studies on mice lacking Kcc3 have demonstrated a normal-appearing corpus callosum, suggesting that Kcc3 may not play a role in either embryonic or postnatal formation of the corpus callosum in mice [Shekarabi et al 2012].
Mechanism of disease causation. Loss of function
Table 7.
Notable SLC12A6 Pathogenic Variants
Chapter Notes
Acknowledgments
This work was supported by La Fondation des Jumelles Coudé and the Canadian Institute of Health Research.
Additionally, the authors would like to acknowledge the very significant contributions of Dr Jean Mathieu and Dr Jean-Pierre Bouchard to the understanding of HMSN/ACC, which have been the cornerstone of its phenotypic and genotypic characterization in the last decade.
Author History
Jean-Denis Brisson, MD, FRCP(C) (2020-present)
Nicolas Dupré, MD, MSc, FRCP(C) (2006-present)
Claudie Gauvreau, MD, MSc (2020-present)
Heidi C Howard, PhD; Radboud University Medical Center (2006-2020)
Guy A Rouleau, MD, PhD, FRCP(C); McGill University (2006-2020)
Revision History
- 17 September 2020 (bp) Comprehensive update posted live
- 12 June 2014 (me) Comprehensive update posted live
- 18 June 2009 (me) Comprehensive update posted live
- 2 February 2006 (me) Review posted live
- 2 September 2005 (gr) Original submission
References
Literature Cited
- Antoniadi T, Buxton C, Dennis G, Forrester N, Smith D, Lunt P, Burton-Jones S. Application of targeted multi-gene panel testing for the diagnosis of inherited peripheral neuropathy provides a high diagnostic yield with unexpected phenotype-genotype variability. BMC Med Genet. 2015;16:84. [PMC free article: PMC4578331] [PubMed: 26392352]
- Auer RN, Laganière J, Robitaille Y, Richardson J, Dion PA, Rouleau GA, Shekarabi M. KCC3 axonopathy: neuropathological features in the central and peripheral nervous system. Mod Pathol. 2016;29:962–76. [PubMed: 27230413]
- Delpire E, Kahle KT. The KCC3 cotransporter as a therapeutic target for peripheral neuropathy. Expert Opin Ther Targets. 2017;21:113–6. [PubMed: 28019725]
- Dupré N, Howard HC, Mathieu J, Karpati G, Vanasse M, Bouchard JP, Carpenter S, Rouleau GA. Hereditary motor and sensory neuropathy with agenesis of the corpus callosum. Ann Neurol. 2003;54:9–18. [PubMed: 12838516]
- Flores B, Schornak CC, Delpire E. A role for kcc3 in maintaining cell volume of peripheral nerve fibers. Neurochemistry international. 2019;123:114–24. [PMC free article: PMC6398598] [PubMed: 29366908]
- Howard HC, Mount DB, Rochefort D, Byun N, Dupré N, Lu J, Fan X, Song L, Rivière JB, Prévost C, Horst J, Simonati A, Lemcke B, Welch R, England R, Zhan FQ, Mercado A, Siesser WB, George AL Jr, McDonald MP, Bouchard JP, Mathieu J, Delpire E, Rouleau GA. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet. 2002;32:384–92. [PubMed: 12368912]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Larbrisseau L, Sarnat H. Andermann syndrome. In: Roos RP, ed. MedLink Neurology. San Diego: MedLink Corporation; 2017.
- Larbrisseau A, Vanasse M, Brochu P, Jasmin G. The Andermann syndrome: agenesis of the corpus callosum associated with mental retardation and progressive sensorimotor neuronopathy. Can J Neurol Sci. 1984;11:257–61. [PubMed: 6329500]
- Mathieu J, Bédard F, Prévost C, Langevin P. Motor and sensory neuropathies with or without agenesis of the corpus callosum: a radiological study of 64 cases. Can J Neurol Sci. 1990;17:103–8. [PubMed: 2357646]
- Salin-Cantegrel A, Rivière JB, Dupré N, Charron FM, Shekarabi M, Karéméra L, Gaspar C, Horst J, Tekin M, Deda G, Krause A, Lippert MM, Willemsen MA, Jarrar R, Lapointe JY, Rouleau GA. Distal truncation of KCC3 in non-French Canadian HMSN/ACC families. Neurology. 2007;69:1350–5. [PubMed: 17893295]
- Shekarabi M, Moldrich RX, Rasheed S, Salin-Cantegrel A, Laganière J, Rochefort D, Hince P, Huot K, Gaudet R, Kurniawan N, Sotocinal SG, Ritchie J, Dion PA, Mogil JS, Richards LJ, Rouleau GA. Loss of neuronal potassium/chloride cotransporter 3 (KCC3) is responsible for the degenerative phenotype in a conditional mouse model of hereditary motor and sensory neuropathy associated with agenesis of the corpus callosum. J Neurosci. 2012;32:3865–76. [PMC free article: PMC6703451] [PubMed: 22423107]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Sun YT, Tzeng SF, Lin TS, Hsu KS, Delpire E, Shen MR. Kcc3 deficiency-induced disruption of paranodal loops and impairment of axonal excitability in the peripheral nervous system. Neuroscience. 2016;335:91–102. [PubMed: 27568057]
- Uyanik G, Elcioglu N, Penzien J, Gross C, Yilmaz Y, Olmez A, Demir E, Wahl D, Scheglmann K, Winner B, Bogdahn U, Topaloglu H, Hehr U, Winkler J. Novel truncating and missense mutations of the KCC3 gene associated with Andermann syndrome. Neurology. 2006;66:1044–8. [PubMed: 16606917]
Publication Details
Author Information and Affiliations
Laval University
Quebec City, Canada
Centre Intégré Universitaire de Santé et de Services Sociaux du Saguenay-Lac-Saint-Jean;
Sherbrooke University
Quebec, Canada
Department of Neurological Sciences
CHA – Enfant-Jésus
Quebec City, Canada
Publication History
Initial Posting: February 2, 2006; Last Update: September 17, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Gauvreau C, Brisson JD, Dupré N. Hereditary Motor and Sensory Neuropathy with Agenesis of the Corpus Callosum. 2006 Feb 2 [Updated 2020 Sep 17]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.