Clinical Description
PIK3CA-related overgrowth spectrum (PROS) includes overgrowth of a broad range of tissues that may or may not be accompanied by cellular dysplasia. Prior to the understanding of the molecular nature of PROS, a number of distinct but overlapping phenotypes were clinically described and given names (see GeneReview Scope).
In general, PROS can be divided into an isolated form (when a person has a focal lesion that affects only one tissue or body part; see Table 2) and a syndromic form (i.e., overgrowth plus at least two other features in two systems; see Table 3). A targeted therapy aimed at inhibiting PI3K-related pathway overgrowth has been approved by the FDA (see Table 6).
Table 2.
Selected Isolated PIK3CA-Related Overgrowth Phenotypes by Affected Organ or Tissue
View in own window
| Organ or Tissue | Phenotype | Comment |
|---|
|
Brain/Head
| HMEG: brain overgrowth affecting 1 hemisphere w/or w/o cortical dysplasia | Cognitive & developmental disabilities Seizures are common. Focal neurologic deficits may be present. May result in facial asymmetry
|
| Focal cortical dysplasia 1 | Types I, II, III 2 Overgrowth not as exaggerated as in HMEG May → epilepsy, which may be refractory to medications if childhood onset Cognitive impairment
|
| DMEG: focal brain overgrowth w/cortical dysplasia; cortical dysplasia may be bilateral. |
|
| Facial infiltrating lipomatosis | Unilateral hypertrophy of soft tissues of the face (most commonly cheek) w/underlying fat infiltration 4, 5 May incl bony hypertrophy
|
|
Limb
| Hemihyperplasia | May incl whole limb, part of limb, or only hand or foot (acral overgrowth) May involve soft tissue, muscle, &/or bone
|
| Macrodactyly |
|
| Fibroadipose vascular anomaly | Fibrofatty tissue w/dilated veins (phlebectasia) replaces muscle fibers. Low-flow venous or lymphatic malformation 6 May present as painful lump, typically of the gastrocnemius May be assoc w/ankle dorsiflexion & calf contracture
|
| Macrodactyly or macrodystrophia lipomatoma | Fibroadipose tissue enlargement & bony overgrowth w/in a nerve territory (e.g., upper or lower limbs, hands & feet) w/↑ length & circumference of peripheral nerve Growth can be static (proportionate) or progressive (disproportionate).
|
|
Lymphatics 7
| Isolated lymphatic malformations: dilated vascular channels lined by lymphatic endothelial cells | Fluid-filled cysts usually grow proportionally w/growth of affected person; may → pain &/or morbidity if they are infiltrative. |
|
Vascular 7
| Vascular malformations | Incl capillary, venous, or mixed malformations |
|
Skin
|
| Skin lesions are typically benign. |
Overgrowth can affect any part of the body depending on the distribution of the PIK3CA pathogenic variant in various tissues and organs.
DMEG = dysplastic megalencephaly; HMEG = hemimegalencephaly
- 1.
Often characterized histologically as having dysplastic neurons, balloon cells, and lamination disorganization
- 2.
Type I: isolated focal lesions with architectural abnormalities
Type II: isolated focal lesions with architectural and dysmorphic abnormalities
Type III: cortical disorganization associated with or adjacent to other principal lesions
- 3.
Seizures are typically partial and may include infantile spasms, tonic seizures, or electroclinical features of Ohtahara syndrome.
- 4.
May be associated with precocious dental development, macrodontia, hemimacroglossia, protuberances on the tongue and buccal mucosa, and mucosal neuromas
- 5.
- 6.
Rare high-flow variants due to excessively muscularized venous channels. Organizing thrombi may be present.
- 7.
Overgrowth of capillary, lymphatic, and venous channels is sometimes referred to as CLVM (capillary lymphatic venous malformations), in which dilated lymphatic channels are combined with venous and capillary components.
Table 3.
Selected Syndromic PIK3CA-Related Overgrowth Phenotypes
View in own window
| Phenotype 1 | Types of Overgrowth | Malformations/Abnormalities |
|---|
| Cutaneous & vascular | Musculoskeletal | Visceral | Neurologic |
|---|
| CLOVES (See .) |
|
|
|
|
|
| CLAPO | Partial/generalized of soft tissues & bone |
| | | |
| FH or FAO | Segmental & progressive overgrowth of subcutaneous & visceral fibroadipose tissue Occasional skeletal overgrowth Disproportionate linear overgrowth
|
|
|
| |
| HHML |
| Multiple lipomas | | | |
| KTS | Bone &/or soft tissue overgrowth in a unilateral limb |
| Digital enlargement | | |
| MCAP or M-CM (See , .) |
| Cutaneous vascular malformations, esp cutis marmorata & capillary malformations of the face |
| Wilms tumor (rare) |
|
| MPPH 3 | Brain overgrowth 4 | | Syndactyly Depressed nasal bridge
| |
|
CLAPO = capillary malformation of the lower lip, lymphatic malformation of the face and neck, asymmetry and partial/generalized overgrowth; CLOVES = congenital lipomatous overgrowth, vascular malformations, epidermal nevi, scoliosis/skeletal and spinal; DD = developmental delay; EN = epidermal nevi; FH or FAO = fibroadipose hyperplasia or overgrowth; HHML = hemihyperplasia multiple lipomatosis; HMEG = hemimegalencephaly; ID = intellectual disability; KTS = Klippel-Trenaunay syndrome; MCAP or M-CM = megalencephaly-capillary malformation; MPPH = megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome
- 1.
Most common findings; see text for more information.
- 2.
May also include cortical dysplasia, polymicrogyria, Arnold-Chiari malformation, and ventriculomegaly
- 3.
Individuals with this phenotype may also display dysmorphic features including prominent forehead, widely spaced eyes, downslanted palpebral fissures, low-set ears, preauricular pits, anteverted nares, and a high and narrow palate.
- 4.
May include megalencephaly, hydrocephalus, and polymicrogyria
Overgrowth
PIK3CA pathogenic gain-of-function variants can cause overgrowth of almost any type of tissue. Onset of overgrowth is typically congenital or early postnatal, in contrast to onset in later infancy or childhood.
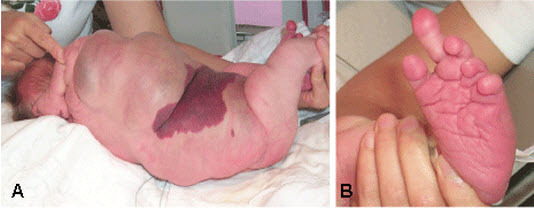
The predominant areas of involvement include the brain, limbs (including fingers and toes), trunk (including abdomen and chest), and face, all usually in an asymmetric distribution.
Overgrown tissue may have a "ballooning" appearance ‒ that is, the involved body part (usually finger/s, toe/s, and/or dorsum of the hand or foot) resembles an inflated balloon.
Unilateral involvement is more common than bilateral involvement.
Overgrowth may include some or all of the following tissue types:
Fibrous, including dense fibrous tissue encircling the nerves in individuals with the fibroadipose vascular anomaly (See
Table 2.)
Brain Growth
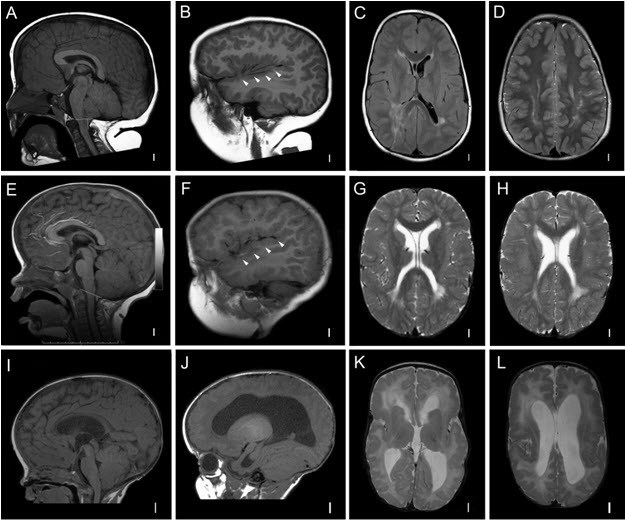
Generalized brain overgrowth may be accompanied by secondary overgrowth of specific brain structures resulting in ventriculomegaly, a markedly thick corpus callosum, and cerebellar tonsillar ectopia with crowding of the posterior fossa [Conway et al 2007b, Mirzaa et al 2012]. Adult OFCs range from +2 to as large as +10 SDs above the mean.
Megalencephalopathy. Although most affected individuals have macrocephaly due to megalencephaly (MEG) at birth, a few affected individuals have normal head size at birth but develop progressive macrocephaly due to progressive MEG within the first year of life [Moore et al 1997, Conway et al 2007a, Conway et al 2007b, Mirzaa et al 2012].
In children who have undergone neurosurgical shunting for obstructive ventriculomegaly or hydrocephalus, head growth noticeably continues at an accelerated pace, indicating the primary nature of MEG in individuals who have MEG as part of their PROS findings.
MCAP syndrome. In a review of 21 children, birth occipitofrontal circumference (OFC) typically ranged from +2 to +7 SDs above the mean for gestational age [Mirzaa et al 2012; Author, unpublished data].
In most children, OFC SD increased during the first year of life. Although head growth may level off in early childhood, it typically remains at +3 SD or more above the mean.
Skeletal Findings
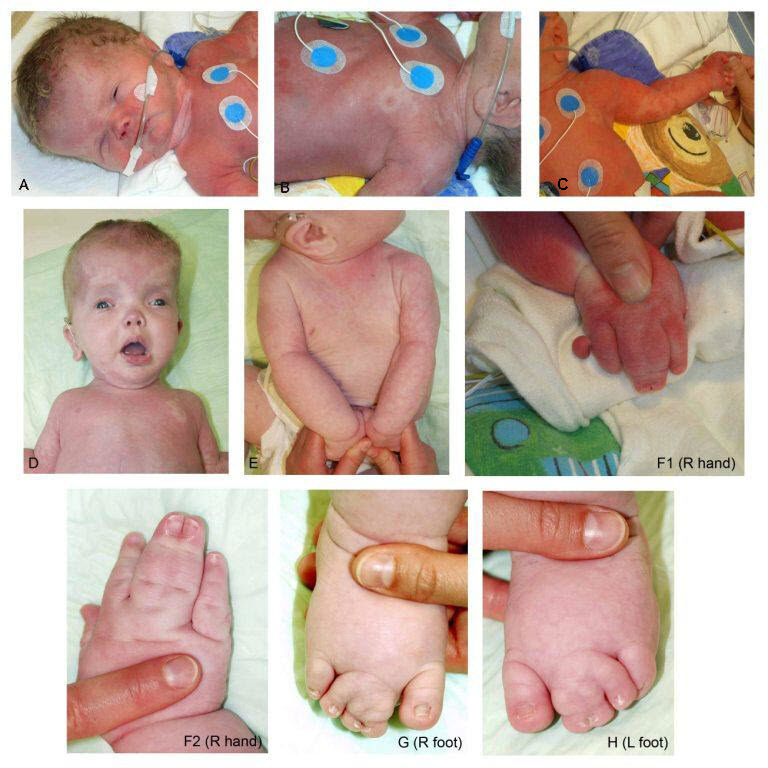
Characteristic findings in the hands include broad, spade-like hands with splayed or ulnar deviation of the fingers and overgrowth of one or more fingers.
Characteristic findings in the feet include overgrowth with a large "sandal" gap between the great and second toes, large bulbous toes, lipomatous masses on both the dorsal and plantar surfaces, or broad forefoot with wide gaps between the metatarsal heads.
Patterning defects may include postaxial, preaxial, or central polydactyly and cutaneous syndactyly, which often involves the toes, but can include the fingers. The cutaneous syndactyly occurs in patterns of 2-3 toes, 2-4 toes, and 2-5 toes with sandal-gap toes.
Dislocated knees, leg-length discrepancy, and pattern chondromalacia can occur.
Some affected individuals may have scoliosis, vertebral anomalies, spina bifida, and/or pectus anomalies, particularly in the CLOVES phenotype.
Progressive skeletal overgrowth has been described in those with the FH or FAO phenotype.
Individuals with the MCAP phenotype may have joint hypermobility due to connective tissue dysplasia.
Lipomatous Overgrowth with or without Regional Reduction of Adipose Tissue
Lipomatous overgrowth may occur ipsilateral or contralateral to a vascular malformation, if present. The characteristic truncal lipomatous mass infiltrates surrounding tissues and often requires surgical excision. Severe scoliosis, large truncal mass, paraspinal high-flow lesions with spinal cord ischemia, lymphatic malformations, cutaneous vesicles, orthopedic problems of the feet and hands, and central phlebectasia/thromboembolism are examples of significant morbidities that need active or prophylactic medical intervention (see Management).
Paraspinal and intraspinal extension, more commonly seen in individuals with the CLOVES phenotype, present significant risk for compression of the cord, thecal sac, and nerve roots, with resultant major neurologic deficits including myelopathy, warranting prompt diagnosis and multidisciplinary care [
Alomari 2009].
Lipomatosis can be invasive, invading hip joints and intravertebral spaces, which can become quite painful.
Infiltration of adipose tissue into muscle with either replacement or compression of muscle, as well as into viscera (liver, spleen, pancreas), intestines, mediastinum, and spine has also been described.
Surgery can be difficult because of the vascularity of the lipomatous tissue and the risk of thrombosis.
PIK3CA pathogenic gain-of-function variants can cause regional reduction of adipose tissue that is often accompanied by significant overgrowth in another part of the body. For example, reduction of adipose tissue in the upper limbs, chest, or upper abdomen has been observed in those with significant overgrowth in the lower body or lower limbs/feet.
Developmental Delay and Intellectual Disability
The degree of intellectual disability (ID) appears to be mostly related to the presence and severity of seizures, cortical dysplasia (e.g., polymicrogyria), and hydrocephalus (see Neuroimaging). Gross motor delays are probably attributable to multiple factors in the affected individual including the presence of MEG, cortical brain malformations, hypotonia, limb asymmetry or overgrowth, and connective tissue dysplasia.
MCAP syndrome
Most individuals with MCAP syndrome have some intellectual disability; the degree is variable and ranges from mild learning disability to severe disability.
Most have mild-moderate delays yet continue to make steady developmental progress, albeit at a slower rate.
A few (<10%) have severe handicaps. The range of expected milestone acquisition has not yet been clarified in individuals with MCAP syndrome.
Other Neurodevelopmental Features
Hypotonia. Younger children with polymicrogyria (specifically of the frontal region) have hypotonia, are not spastic, and may have pseudobulbar problems; older children often have spasticity and pseudobulbar problems [Mirzaa et al 2012].
Infant feeding difficulties. Many children may have feeding difficulties that are often multifactorial in nature (e.g., due to hypotonia, GERD), although use of a feeding tube is rarely required.
Epilepsy. An estimated 30%-40% of individuals with PIK3CA pathogenic variants have epilepsy. Reported seizure types include focal and tonic-clonic (among others), though seizure types, severity, age of onset, and any associated EEG or neuroimaging abnormalities depend primarily on the tissue distribution of the pathogenic PIK3CA variants and the presence or absence of associated cortical malformations.
Symptoms of cerebellar tonsillar ectopia (Chiari malformation). Infants may have irritability, excessive drooling, difficulty swallowing, or breathing problems, especially central apnea. Children may have neck pain or headache, motor weakness, sensory changes, vision problems, swallowing difficulties, or behavioral changes.
Behavioral Issues and Autistic Features
A subset of children (6/21) with MCAP syndrome have autistic features or a clinical diagnosis of autism [Mirzaa et al 2012], suggesting that autism may be part of the neurocognitive profile of a minority of individuals who have MEG as part of PROS [McBride et al 2010]. Other behavioral abnormalities seen in one or a few affected individuals:
Neuroimaging
Individuals with PROS who have macrocephaly typically undergo brain imaging shortly after birth or within the first year of life, leading to early identification of the following key neuroimaging features (see ) [Vogels et al 1998, Nyberg et al 2005, Conway et al 2007a, Conway et al 2007b, Martínez-Lage et al 2010, Mirzaa et al 2012].
Megalencephaly [Clayton-Smith et al 1997, Vogels et al 1998, Robertson et al 2000, Nyberg et al 2005, Coste et al 2012]. In some instances, MEG (with or without ventriculomegaly) is detected prenatally on ultrasound examination, along with a thickened corpus callosum. More than 90% of affected individuals have congenital MEG that is universally progressive.
Ventriculomegaly and hydrocephalus. Most affected children have evidence of ventricular dilatation or ventriculomegaly on early brain imaging:
In a large review of the neuroimaging findings in individuals with MCAP syndrome, 37 (56%) of 65 children had ventriculomegaly ranging from mild-to-frank hydrocephalus, with or without cerebellar tonsillar ectopia [
Conway et al 2007b].
While it is unclear whether ventriculomegaly is obstructive in all these individuals, more than half of affected children undergo ventricular shunting or third ventriculostomy, usually within the first year of life.
Cerebellar tonsillar ectopia (CBTE). A large cerebellum combined with a small posterior fossa leading to cerebellar tonsillar ectopia (Chiari malformation) and syringomyelia are common complications, particularly in those with MCAP syndrome:
Fifteen of 65 reported affected individuals with MCAP have evidence of CBTE with or without herniation [
Conway et al 2007b].
The degree of ectopia is best objectively assessed by measuring the distance of the cerebellar tonsils below the foramen magnum.
Unlike ventriculomegaly, CBTE is rarely congenital in individuals with MCAP syndrome.
In two individuals spontaneous "resolution" of CBTE on follow-up imaging was attributed to disproportionately accelerated skull overgrowth [
Mirzaa et al 2012].
Cortical brain malformation and polymicrogyria (PMG). PMG may be present in more than 50% of affected children, most commonly those with the MCAP phenotype [Conway et al 2007b; Gripp et al 2009; Mirzaa et al 2012; Authors, unpublished data].
The most common type of PMG in those with MCAP syndrome is bilateral perisylvian PMG, although other types including bilateral frontal and focal PMG occur.
PMG broadly, and bilateral perisylvian PMG in particular, increase the risk for: epilepsy; oral motor weakness leading to feeding, swallowing, and expressive language difficulties; developmental delay; and tone abnormalities.
Tumors
Benign tumors. The most common are vascular, described variably as (cavernous) hemangiomas, angiomata, angiomyolipomas, and vascular masses [Clayton-Smith et al 1997, Moore et al 1997, Martínez-Glez et al 2010].
Cavernous hemangiomas have occurred in brain tissue, necessitating debulking when they enlarge and cause pain.
While most common in skin or subcutaneous tissue, hemangiomas have been described in viscera and skull.
Other benign tumors have included two individuals with MCAP (ages 21 months and 5 years) who had meningioma (which do not tend to enlarge, spread, or metastasize); two individuals with PROS who had spinal and major nerve neurofibromas; and several others with ovarian cystadenoma, uterine fibroids, and lipomas [Keppler-Noreuil et al 2014].
Malignant tumors. If present, most affected individuals have benign tumors, with only a few malignant tumors reported [Kurek et al 2012, Keppler-Noreuil et al 2014, Luks et al 2015, Gripp et al 2016, Hucthagowder et al 2017, Kuentz et al 2017, Peterman et al 2017, Postema et al 2017].
The estimated frequency of Wilms tumor ranges from 1.4% to 3.3%. Of 12 individuals reported to have Wilms tumor or nephroblastomatosis, clinical PROS diagnoses included CLOVES (8 individuals), MCAP (2 individuals), and KTS (2 individuals).
Mean age at diagnosis was 27.4 months (median 18 months; range 9-119 months).
Tumor type included seven individuals (~60%) with Wilms tumor, four (33%) with indeterminate features of Wilms tumor vs nephroblastomatosis, and one (8%) with nephroblastomatosis.
There have been several case reports of individuals with PROS who developed other cancers including the following [Moore et al 1997, Schwartz et al 2002, Mills et al 2018]:
Systematic data are at present insufficient to determine whether there is a true association between PROS and the development of these types of tumors or whether the case reports represent rare co-occurrences of PROS with these tumors.
Other
Kidney. Kidney malformations are frequently found in individuals with PROS, and more specifically the CLOVES phenotype, and include pelviectasis, dilated ureters, hydronephrosis, duplicated renal arteries, renal cysts, and enlarged kidneys.
Skin. Abnormalities observed in PROS:
Dermal melanocytic nevi
Café au lait macules
Hypopigmented macules
Cutis marmorata
Pigmented nevi
Patchy hyperpigmentation that follows the lines of Blaschko
Linear keratinocytic epidermal nevi, which may occur anywhere on the body and may follow a dermatomal distribution
Seborrheic keratosis and benign lichenoid dermatosis
Skin hyperelasticity, laxity, and thick subcutaneous tissue in those with the MCAP phenotype due to connective tissue dysplasia
Endocrine issues affect a small number of individuals and most commonly include hypoglycemia (largely hypoinsulinemic hypoketotic hypoglycemia), growth hormone deficiency, and hypothyroidism [Mirzaa et al 2016, Leiter et al 2017, Davis et al 2020, Maines et al 2021, Douzgou et al 2022].
PROS is a clinical phenocopy of congenital hyperinsulinism (see
Molecular Genetics), but plasma insulin concentrations at the time of hypoglycemia are undetectable.
This profile has been reported both in individuals with MCAP and in those with overlapping forms of PROS that include brain involvement.
While hypoglycemia in PROS is most commonly diagnosed in the neonatal period, some individuals may present later in childhood, including at least one male with MCAP who presented with his first episode of hypoglycemia at age six years [Author, personal communication].
Hypoglycemia may be persistent over time, requiring consistent glucose monitoring, evaluation of the hypothalamic-pituitary-adrenal axis, and ongoing treatment (see
Management).
Cardiac issues. Structural heart defects (e.g., atrial and ventricular septal defects) and/or abnormalities of the great vessels have been reported in some individuals with MCAP syndrome [Mirzaa et al 2016].