Clinical characteristics.
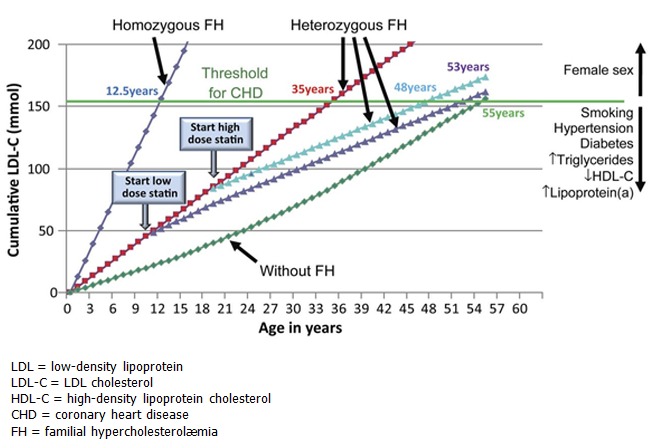
Familial hypercholesterolemia (FH) is characterized by significantly elevated low-density lipoprotein cholesterol (LDL-C) that leads to atherosclerotic plaque deposition in the coronary arteries and proximal aorta at an early age and increases the risk of premature cardiovascular events such as angina and myocardial infarction; stroke occurs more rarely. Xanthomas (cholesterol deposits in tendons) may be visible in the Achilles tendons or tendons of the hands and worsen with age as a result of extremely high cholesterol levels. Xanthelasmas (yellowish, waxy deposits) can occur around the eyelids. Individuals with FH may develop corneal arcus (white, gray, or blue opaque ring in the corneal margin as a result of cholesterol deposition) at a younger age than those without FH.
Individuals with a more severe phenotype, often as a result of biallelic variants, can present with very significant elevations in LDL-C (>500 mg/dL), early-onset coronary artery disease (CAD; presenting as early as childhood in some), and calcific aortic valve disease.
Diagnosis/testing.
A clinical diagnosis of FH can be established in a proband with characteristic clinical features and significantly elevated LDL-C levels (typically >190 mg/dL in adults and >160 mg/dL in children). Three formal diagnostic criteria are used in Western countries.
The molecular diagnosis of FH can be established by identification of heterozygous or biallelic pathogenic variants in APOB (variants that impair binding of LDL-C to the LDL receptor), LDLR, or PCSK9 (gain of function); or rarely, identification of biallelic pathogenic variants in LDLRAP1.
Management.
Treatment of manifestations: Adults: pharmacotherapy (statins with additional medications as needed) to reduce lipid levels; referral to a lipid specialist if necessary to reduce LDL-C levels; reduce CAD risk factors including cessation of smoking, regular physical activity, healthy diet, and weight control; treatment of hypertension; low-dose aspirin in high-risk individuals. Children: referral to a lipid specialist; diet and lifestyle modifications; statins can be used in children starting around age eight years.
Prevention of primary manifestations: Heart-healthy diet (including reduced intake of saturated fat and increased intake of soluble fiber to 10-20 g/day); increased physical activity; no smoking.
Surveillance: Monitor lipid levels from age two years; consider noninvasive imaging modalities in adults; identify modifiable risk factors (e.g., smoking, sedentary behavior, hypertension, diabetes, obesity). Individuals with severe FH (i.e., due to homozygous or compound heterozygous pathogenic variants in APOB, LDLR, or PCSK9) or autosomal recessive FH (due to homozygous or compound heterozygous pathogenic variants in LDLRAP1) should be monitored with various imaging modalities (including echocardiogram, CT angiogram, and cardiac catheterization) as recommended.
Agents/circumstances to avoid: Smoking, high intake of saturated and trans unsaturated fat, sedentary lifestyle, obesity, hypertension, and diabetes mellitus.
Evaluation of relatives at risk: Early diagnosis and treatment of first-degree and second-degree relatives at risk for FH can reduce morbidity and mortality. The genetic status of at-risk family members can be clarified by either: (1) molecular genetic testing if the pathogenic variant(s) has been identified in an affected family member; or (2) measurement of LDL-C concentration. Genetic testing is the preferred method for clarifying the diagnosis in at-risk family members, when possible.
Pregnancy management: Pregnant women should incorporate all the recommended lifestyle changes, including low saturated fat intake, no smoking, and high dietary soluble fiber. Statins are contraindicated in pregnancy because of concerns for teratogenicity and should be discontinued prior to conception. Bile acid-binding resins (e.g., colesevelam) are generally considered safe (Class B for pregnancy), and LDL apheresis is also used occasionally if there is evidence of established CAD. Use of PCSK9 inhibitors, ezetimibe, lomitapide, and bempedoic acid during pregnancy has not been well studied.
Genetic counseling.
APOB-, LDLR-, and PCSK9-related FH are inherited in an autosomal dominant manner. If an individual has biallelic (homozygous or compound heterozygous) pathogenic variants in one of these three genes – a condition referred to as homozygous FH (HoFH) – the presentation becomes more severe with earlier onset of features. Some individuals with FH are heterozygous for pathogenic variants in two different FH-related genes, which may have an additive effect on the severity of FH. Each child of an individual with a heterozygous pathogenic variant in APOB, LDLR, or PCSK9 has a 50% chance of inheriting the pathogenic variant and having FH. All children of an individual with homozygous FH will inherit a pathogenic variant and have FH. If the reproductive partner of a proband is heterozygous for an FH-related pathogenic variant in the same gene as the proband or a different FH-related gene, offspring are at risk of inheriting two pathogenic variants and having severe FH.
LDLRAP1-related FH is caused by biallelic pathogenic variants and is inherited in an autosomal recessive manner. The parents of an individual with LDLRAP1-related FH are presumed to be heterozygous for one pathogenic variant. If both parents are known to be heterozygous for an LDLRAP1 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being a carrier, and a 25% chance of inheriting neither of the familial pathogenic variants. Carrier testing for at-risk relatives requires prior identification of the LDLRAP1 pathogenic variants in the family.
Once the FH-causing pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.