Summary
Clinical characteristics.
Nicolaides-Baraitser syndrome (NCBRS) is characterized by sparse scalp hair, prominence of the inter-phalangeal joints and distal phalanges due to decreased subcutaneous fat, characteristic coarse facial features, microcephaly, seizures, and developmental delay / intellectual disability. Seizures are of various types and can be difficult to manage. Developmental delay / intellectual disability (ID) is severe in nearly a half, moderate in a third, and mild in the remainder. Nearly a third never develop speech or language skills.
Diagnosis/testing.
The diagnosis of NCBRS is established in a proband with suggestive clinical findings and the identification of a heterozygous SMARCA2 pathogenic variant by molecular genetic testing.
Management.
Treatment of manifestations: Anti-seizure medication (ASM) for seizures under the care of a neurologist or epileptologist; occupational, physical, and/or speech therapy; routine management of refractive errors and hearing loss.
Surveillance: At least yearly evaluation by a neurologist to assess for and/or manage seizures; yearly evaluation by a developmental pediatrician to assess developmental progress and therapeutic and educational interventions; regular follow up of ophthalmologic and/or audiologic abnormalities.
Genetic counseling.
NCBRS is inherited in an autosomal dominant manner. All affected individuals reported to date have NCBRS as the result of a de novo
SMARCA2 pathogenic variant. Although no affected sibs have been reported, the risk to sibs is presumed to be greater than in the general population because of the possibility of germline mosaicism in a parent. If the SMARCA2 pathogenic variant has been identified in an affected family member, prenatal testing for pregnancies at theoretic increased risk is possible.
Clinical Characteristics
Clinical Description
The most common findings in 61 individuals (35 male and 26 female) with Nicolaides-Baraitser syndrome (NCBRS) and a SMARCA2 pathogenic variant are summarized in Table 2 [Sousa et al 2014].
Table 2.
Summary of the Most Common Clinical Findings in 61 Individuals with Nicolaides-Baraitser Syndrome
View in own window
| Finding | % of Affected Individuals |
|---|
| Intellectual disability | 100% |
| Sparse hair | 97% |
| Prominent interphalangeal joints | 85% |
| Coarse facies | 77% |
| Microcephaly | 65% |
| Seizures | 64% |
Ectoderm. Scalp hair is usually sparse at birth and becomes increasingly so with age, particularly in the second decade of life. In some, the sparseness improves with time.
Skin pigmentation appears to be reduced, although affected individuals do not exhibit true cutaneous albinism.
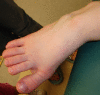
Poor subcutaneous fat distribution leads to prominent veins; interphalangeal joints are prominent. The distal phalanges widen with age, becoming oval shaped and broad. Increasing space between the first and second toes can occur over time.
Delayed tooth eruption is common, often requiring surgical extraction of primary dentition to allow secondary dentition to migrate into place.
Seizures can be difficult to manage, requiring multiple anti-seizure medications to achieve reasonable control. The mean and median age of onset of seizures is 24 months and 18 months, respectively. The range is birth to 14 years. Various seizure types have been reported. Regression or lack of developmental progress has been noted with the onset of seizures.
Developmental delay / intellectual disability. Nearly half of affected individuals experience severe developmental delay / intellectual disability (ID) with particular delays in speech and language development. A third exhibit moderate intellectual disability and the remainder have mild ID.
Nearly a third never develop speech or language skills.
The mean age for walking independently is 21 months (range: 10 months - 5 years).
Although psychomotor regression is not typical, the high incidence of seizures that progressively worsen has been associated with loss of speech.
Behavior problems have been reported in at least 19 individuals with some displaying autistic-like features (such as perseveration and hyperacusis). However, a formal clinical diagnosis of autism has not been made in any affected individual reported in the literature.
Growth. About half of affected individuals (24/46) have low birth weight and the same proportion experience short stature. None was above the 50th centile for height.
Microcephaly tends to be acquired, being noted in almost one third of infants at birth (7/30) and in two thirds (34/52) at follow up.
Other findings
Cryptorchidism is seen in most males.
Hearing loss has been noted in 4/59 affected individuals. The nature of the hearing loss and age of onset have not been reported.
Visual deficits including myopia and astigmatism have been seen in ten and four affected individuals, respectively.
Congenital heart defects (e.g., atrial septal defect, stenosis of the pulmonary artery, coarctation, patient ductus arteriosus, and double aortic arch) have been reported in six affected persons.
Genotype-Phenotype Correlations
No clear genotype-phenotype correlations have been noted; however, all individuals with a pathogenic variant within the C-terminal helicase region of the ATPase domain have severe intellectual disability and epilepsy – a frequency higher than that in individuals with pathogenic variants in other parts of the gene.
Penetrance
Data are insufficient to determine penetrance. All 61 affected individuals published to date had a de novo pathogenic variant, suggesting that penetrance is likely complete [Sousa et al 2014].
Nomenclature
Morin et al [2003] proposed the name Nicolaides-Baraitser syndrome after the authors of the 1993 article in which the earliest known person with NCBRS was described.
Prevalence
The prevalence of NCBRS is not known, but is estimated to be extremely low. Fewer than 100 affected individuals have been described in the literature.
Differential Diagnosis
Coffin-Siris syndrome
(CSS) is characterized by coarse facial features, hypertrichosis, absent or hypoplastic fifth nails (fingers and/or toes), agenesis of the corpus callosum, and developmental delay / intellectual disability. Although the facial features can be similar to those of Nicolaides-Baraitser syndrome (NCBRS), the digital findings are particularly helpful in differentiating the two disorders, as individuals with NCBRS uniquely have prominent interphalangeal joints and do not have hypoplasia of the fifth digits. Additionally, individuals with NCBRS have sparse scalp hair rather than the hypertrichosis seen in CSS.
CSS is caused by a heterozygous pathogenic variant in one of the following genes: ARID1A, ARID1B (see ARID1B-Related Disorder), SMARCA4, SMARCB1, or SMARCE1. In the majority of affected individuals the pathogenic variant is de novo.
The phenotypic overlap between CSS and NCBRS is likely due to the fact that both conditions are caused by pathogenic variants in genes involved in the SWI/SNF complex.
Of note, Tsurusaki et al [2012] reported an individual with a clinical diagnosis of Coffin-Siris syndrome (CSS) who had a 55-kb interstitial deletion of SMARCA2 and whose clinical findings were more consistent with a diagnosis of NCBRS (prominent interphalangeal joints) than CSS (fifth fingernails were not absent).
Williams syndrome
(WS) is characterized by typical facial features (long philtrum, thick vermilion of the upper and lower lips, and full cheeks), supravalvar aortic stenosis, joint laxity, growth deficiency, hypercalcemia, and intellectual disability. However, their cognitive profile shows a relative strength in language. The facial features, cardiac lesions, and cognitive profile are sufficient to differentiate WS from NCBRS. WS is caused by microdeletion of the Williams-Beuren syndrome critical region (WBSCR). The majority of affected individuals have a de novo microdeletion.
Cornelia de Lange syndrome
(CdLS) is characterized by typical facial features (long philtrum, arched eyebrows, synophrys, and short nose), growth deficiency, and limb anomalies. The limb findings primarily include hypoplasia of the digits and can be severe. The facial features and limb changes are sufficient to differentiate CdLS from NCBRS. CdLS is caused by mutation of one of the following genes: NIPBL, HDAC8, RAD21, SMC1A, or SMC3. In the majority of affected individuals the pathogenic variant is de novo.
2q37 microdeletion syndrome is characterized by typical facial features (round face, frontal bossing, arched eyebrows, upslanted palpebral fissures, epicanthal folds, underdeveloped nasal alae, and thin vermilion of the upper lip), brachymetaphalangy of digits 3-4, short stature, hypotonia, and intellectual disability. Seizures have been reported in individuals with the 2q37 deletion syndrome, but are not usually difficult to manage. In most affected individuals microdeletion is de novo.
Biotinidase deficiency is primarily associated with seizures, hypotonia, and cutaneous abnormalities including hair loss. Facial features are typically normal. Biotinidase deficiency is caused by biallelic pathogenic variants in BTD and is inherited in an autosomal recessive manner.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Nicolaides-Baraitser syndrome (NCBRS) is inherited in an autosomal dominant manner. All affected individuals reported to date have NCBRS as the result of a de novo pathogenic variant.
Risk to Family Members
Parents of a proband
All probands with NCBRS reported to date have the disorder as a result of a de novo
SMARCA2 pathogenic variant.
Recommendations for the evaluation of parents of a proband with an apparent de novo pathogenic variant include testing of the parents for the SMARCA2 pathogenic variant identified in the proband. However, the high penetrance of the disorder suggests that it would be highly unlikely to discover a pathogenic variant in an apparently asymptomatic individual.
Sibs of a proband. To date, all affected individuals have had a de novo
SMARCA2 pathogenic variant, suggesting a low risk to sibs. However, because of the theoretic possibility of parental germline mosaicism, the empiric recurrence risk to sibs is approximately 1%.
Offspring of a proband. Each child of an individual with NCBRS has a 50% chance of inheriting the SMARCA2 pathogenic variant. To date, individuals with NCBRS have not been known to reproduce.
Other family members. The risk to other family members appears to be low given that all probands with NCBRS reported to date have the disorder as a result of a de novo
SMARCA2 pathogenic variant.
Prenatal Testing and Preimplantation Genetic Testing
Once the SMARCA2 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for NCBRS are possible.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Nicolaides-Baraitser Syndrome: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
View in own window
|
600014 | SWI/SNF-RELATED, MATRIX-ASSOCIATED, ACTIN-DEPENDENT REGULATOR OF CHROMATIN, SUBFAMILY A, MEMBER 2; SMARCA2 |
|
601358 | NICOLAIDES-BARAITSER SYNDROME; NCBRS |
Molecular Pathogenesis
The SWI/SNF family of ATPase-dependent chromatin remodelers is essential for the regulation of gene expression, differentiation, and development. SMARCA2 encodes SMARCA2 which is within the family of helicase-related proteins that share an ATPase domain critical for the coupling of ATP hydrolysis with DNA binding, which in turn results in chromatin remodeling. SMARCA2 is a subunit of the BRG1-associated factors (BAF) complex, the human analog of the SWI/SNF complex.
Mutation of other genes within the SWI/SNF complex has been suggested as the cause in individuals who have findings suggestive of Nicolaides-Baraitser syndrome (NCBRS) but no detectable SMARCA2 pathogenic variant [Van Houdt et al 2012].
Gene structure.
SMARCA2 comprises 34 exons and makes a transcript of 5879 base pairs. Alternatively spliced transcript variants encoding different isoforms have been found for this gene, which contains a CAG trinucleotide repeat length polymorphism. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. In a review of 61 individuals with NCBRS, 59 had pathogenic missense variants and two had in-frame deletions [Sousa et al 2014]. All pathogenic variants clustered within the ATPase SMARCA2 domain (exons 15-25). Based on this finding, it is believed that pathogenic variants in SMARCA2 most likely cause a dominant-negative or gain-of-function effect.
Normal gene product.
SMARCA2 codes for a 1,590-amino acid catalytic subunit of the SWI/SNF family of ATPase-dependent chromatin remodelers. The catalytic subunit contains a helicase-like ATPase domain that plays a role in the regulation of gene expression through DNA binding that is coupled with ATP binding and ATP hydrolysis.
Abnormal gene product. Pathogenic variants in SMARCA2 may result in aberrant chromatin remodeling, causing downstream dysregulation of further genes and resulting in the NCBRS phenotype. It is suspected that mutated SMARCA2 is able to incorporate into the SWI/SNF complex that then binds to downstream targets within the genome. In this scenario, remodeling of the nucleosome structure in order to effect gene expression would not be able to occur normally, due to a dominant-negative or gain-of-function effect.