

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Liedtke WB, Heller S, editors. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Boca Raton (FL): CRC Press/Taylor & Francis; 2007.

TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades.
Show detailsAn understanding of the role of TRPV4 in mammalian kidney physiology begins in the nematode C. elegans. Worms are repelled by steep osmotic gradients, an adaptive mechanism presumably conserved to minimize the risk of acute changes in cell volume. Worms with mutated copies of a particular gene, while remaining fully motile, are oblivious to potentially harmful osmotic gradients; this gene was dubbed osm-9 [1,2]. The OSM-9 protein bore a striking similarity to the transient receptor potential channel implicated in visual signal transduction in Drosophila [3]—the prototypical member of the now well-recognized TRP superfamily [4]. Several years later, the mammalian orthologue of OSM-9 was identified via homology screening [5,6] and other approaches [7,8]; it eventually became known as TRPV4 [9].
ACTIVATION OF TRPV4
Activation of TRPV4 by Thermal Stress
Like most other members of the TRPV subfamily, TRPV4 is temperature sensitive. This phenomenon was observed under conditions of both native [10,11] and heterologous [11–13] expression. The role of TRPV4 in thermosensation in vivo, however, is complex. For example, whereas TRPV4-null mice and control mice exhibit similar latencies (time to withdrawal) in response to gradual temperature elevation on a heated plate [14], the null mice prefer a warmer floor temperature than their wild-type littermates do, when presented with a choice [15].
Activation of TRPV4 by Lipids
Although it is unresponsive to the vanilloid TRPV1 agonists capsaicin and resin-iferatoxin [5,6], TRPV4 is activated by a variety of lipids including phorbol 12-myristate 13-acetate (PMA) and 4α-phorbol 12,13-didecanoate (4α-PDD) [16]. This activating effect operates independently of protein kinase C. TRPV4 is also activated by the endogenous cannabinoid anandamide [17], much like TRPV1 (reviewed in reference 18). The anandamide effect is independent of the G-protein-coupled cannabinoid receptors [16,19]. Rather, anandamide is likely metabolized to arachidonic acid and then, via the action of cytochrome P450 epoxygenase, is further metabolized to one of several epoxyeicosatrienoic acid (EET) compounds. These lipids, particularly 5′,6′-epoxyeicosatrienoic acid, then activate TRPV4. Whether activation of TRPV4 by these EETs is direct or indirect remains unresolved. Exogenously applied EETs activate TRPV4 [17]. But there are numerous catabolic pathways for the EETs [20]; much as the TRPV4-activating ability of arachidonic acid was traced to its EET metabolites, so might these putative effects of EET be attributable to one of its many metabolites.
Some lipids, including arachidonic acid and anandamide, indirectly influence the properties of ion channels by altering their lipid microenvironments (see, e.g., references 21 and 22). As discussed earlier, C. elegans mutated for the osm-9 gene (analogous to mammalian TRPV4) fail to avoid osmotic gradients [1,2]. Interestingly, this phenotype is reproduced in worms mutated for a gene encoding a fatty acid desaturase enzyme (fat-3) essential for synthesis of polyunsaturated fatty acids [23]. This sensory defect can be rescued through dietary supplementation of either arachidonic acid or eicosapentaenoic acid [23], suggesting a profound impact of polyunsaturated fatty acids on TRPV4 function.
Mechanosensation and TRPV4
Although TRPV4 is a reputed mechanosensory channel (reviewed in reference 24), the relationship between this role and its osmosensory function (see next section) remains unclear. Data thus far suggest that the channel is only indirectly gated by hypotonicity and that either lipid-mediated or kinase-dependent signaling intermediates are required [17,25,26]. Although it was not activated by direct patch suction, heterologously expressed TRPV4 was variably responsive to positive pressure in the whole-cell configuration [6,27]. In addition, TRPV4 was activated by shear stress from laminar bath flow [13]. In a mechanosensory capacity, it is tempting to speculate that TRPV4 may form part of the primary cilium on kidney tubule epithelial cells; these apical structures sense flow of “proto-urine” in the tubular lumen [28,29]. At least one other TRP family member, polycystin-2, is a constituent of this putative flow-sensing ciliary apparatus. However, immunolocalization studies were inconsistent with this distribution for TRPV4 in the kidney [30]. Interestingly, TRPV4 localized to an analogous ciliary structure in cells lining the oviduct of the female reproductive tract where coexpression with polycystin-2 was also observed [31].
Activation of TRPV4 by Hypotonicity
In heterologous expression systems, TRPV4 is sensitive to minute changes in ambient osmolarity, giving rise to a robust calcium entry signal [5–7]. Conflicting data have emerged concerning the molecular mechanism through which hypotonicity influences TRPV4 function. Xu et al. noted that TRPV4—heterologously expressed in HEK293 cells or natively expressed in a murine kidney distal convoluted tubule cell line—rapidly undergoes tyrosine phosphorylation in response to hypotonic stress, and this phenomenon was sensitive to inhibition of SRC-family cytoplasmic tyrosine kinases [26]. Correspondingly, several SRC family kinases physically associated with TRPV4. Overexpression of one of these kinases, Lyn, enhanced both basal and hypotonicity-inducible tyrosine phosphorylation of TRPV4, whereas overexpression of dominant-negative acting Lyn partially blocked the response. Systematic mutation of tyrosine residues in TRPV4 indicated that Tyr-253 was the site of regulated phosphorylation. Mutation of this site to any of a number of different residues prevented TRPV4-dependent calcium entry in response to hypotonic shock without influencing channel targeting to the cell membrane [26]. The essential nature of this residue was seen in both stable [26] and transient [32] transfection experiments.
Consistent with a role in TRPV4 activation, other data support a role for tyrosine phosphorylation in the cell response to anisotonic stress. Tyrosine phosphorylation of a range of proteins is upregulated by hypotonicity [33–35], and pharmacological manipulation of global tyrosine kinase or phosphatase activity influences the osmo-regulatory efflux of ions and osmotically active organic solutes [34,36,37]. In addition, a number of SRC family cytoplasmic tyrosine kinases are activated by cell swelling and are instrumental in the physiological response to hypotonic stress [38]. Similarly, the SRC family kinase Fyn regulates the tonicity-responsive transcription factor, TonEBP [39]. Moreover, it was later shown that other TRPV channels as well as members of the TRPC and TRPM families are similarly regulated by SRC family cytoplasmic tyrosine kinases [40–44].
In contrast to these findings, Nilius and colleagues concluded that SRC family kinase activity and phosphorylation of Tyr-253 are dispensable for activation of TRPV4 by hypotonicity [25]. In addition to a different expression model, these authors employed single-cell fura-2-based calcium assay in adherent cells, in contrast to a populationwide assessment in suspended cells as Xu et al. had used [26]. The Nilius group further demonstrated via inhibitor studies that phospholipase A2 (PLA2) was essential for TRPV4 activation by hypotonic stress but not by the lipid agonist, 4α-PDD [25]. These data were consistent with earlier findings that hypotonicity activated phospholipase A2-dependent arachidonic acid release [45–47]. Cytochrome P450 epoxygenase—which functions downstream of phospholipase A2 and is required for TRPV4 activation by anandamide—was also a component of the pathway leading to TRPV4 activation by hypotonicity. As an aside, this enzyme complex is not a universal participant among TRPV4-activating pathways; inhibition of P450 epoxygenase prevented calcium entry in response to hypotonic stress but not in response to heat stress [25]. These authors went on to implicate involvement of another tyrosine residue, Tyr-555, in activating TRPV4 by heat and 4α-PDD but, importantly, not by cell swelling [25].
Activation of TRPV4 by Other Stimuli
In addition to responding to heat, lipids, and mechanical stress, TRPV4 might be activated by other physical stimuli. Cilia on the epithelial cells lining the oviduct propel mucus (as well as gametes and embryos) over a wide range of viscosities. When oviduct ciliated cells were superfused in vitro with a viscous medium (supplemented with dextran), an abrupt increase in TRPV4 calcium channel activity was noted [48]. This effect of viscosity was not detectable in naïve HeLa cells, but was recapitulated if the HeLa cells were transfected with TRPV4. Pharmacological inhibition of phospholipase A2 blocked the viscosity-inducible TRPV4-like current in oviduct ciliated cells [48], consistent with a mode of activation for TRPV4 postulated by the Nilius group [25].
TRPV4 IN CELLULAR OSMOREGULATION
Recent data indicate that TRPV4 regulates water balance at the cellular level. Cells subjected to a hypotonic milieu abruptly swell as a consequence of water entry. Unregulated water entry, facilitated by membrane expression of water channel proteins, constitutes a two-pronged threat: the ensuing increase in hydrostatic pressure jeopardizes cell integrity, while dilution of the cytoplasm adversely affects macro-molecular structure and function. Acutely swollen cells jettison ions and organic solutes to permit water efflux in a process known as regulatory volume decrease. This phenomenon has been studied in great detail in a number of in vitro model systems. In most, hypotonic stress is rapidly followed by calcium entry (reviewed in reference 49); this proximal signaling event then triggers efflux of ions (particularly K+ and Cl−) and osmotically active organic solutes such as taurine and sorbitol (reviewed in reference 50).
Although many of the volume-regulatory effector mechanisms triggered by swelling-induced calcium transients are conserved across model systems, the mechanism of calcium entry itself is highly cell type–specific. In the model of human airway epithelial cells, as in most models, the regulatory volume decrease following hypotonic stress is calcium dependent [51]. In this model, both the hypotonicity-dependent calcium entry and subsequent regulatory volume decrease require TRPV4 [52]. Downregulation of endogenous TRPV4 expression with a TRPV4-directed antisense prevented activation of a calcium-dependent potassium channel by hypotonicity and completely prevented restoration of normal cell volume. An irrelevant antisense had no effect (Figure 29.1A). These important data were the first to link TRPV4 function to the regulatory volume decrease response itself, and the data imply a pivotal role for TRPV4 in water balance at the cellular level.
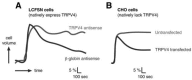
FIGURE 29.1
Role of TRPV4 in cellular osmoregulation. (A) LCFSN cells, which natively express TRPV4, were transfected with antisense directed against TRPV4 or an irrelevant control antisense (beta-globin). Increase in cell volume (percent) was then monitored as a (more...)
Becker and colleagues went on to show a similar relationship [53]. Rather than blocking regulatory volume decrease in TRPV4-expressing cells, they investigated the process in the Chinese hamster ovary (CHO) cell line, which lacks TRPV4 expression. Naïve CHO cells failed to undergo regulatory volume decrease following osmotic swelling (Figure 29.1B); heterologous expression of TRPV4, however, was permissive for this restorative change in cell volume [53]. So it appears that, at least in some models, downregulation of TRPV4 expression abolishes regulatory volume decrease, and heterologous expression of the channel is sufficient to confer this adaptive response.
TRPV4 IN SYSTEMIC OSMOREGULATION
Overview
A physiological role for osmotically responsive TRPV channels is indisputable in C. elegans; among higher eukaryotes, data are emerging more slowly. Initial speculation was fueled by the attractive localization of TRPV4 to key mechanosensory and osmosensory tissues, including the mechanosensitive neurons of the mammalian inner ear hair cells and the cells of the osmosensing blood–brain barrier-deficient circum-ventricular nuclei [5,12]. TRPV4’s expression in the latter context suggested a role in systemic rather than purely cellular osmoregulation. Arguably the best way to assess TRPV4 function in vivo is through targeted gene deletion. Two groups have done this, generating TRPV4−/− mice using slightly but perhaps significantly dissimilar methodologies. Although both sets of knockout mice differ from their wild-type counterparts, they unexpectedly exhibit dissimilar phenotypes (see below). In order to fully appreciate a role for TRPV4 in kidney regulation of water balance, and to speculate on mechanisms through which this might be achieved, it is instructive to review the functional anatomy of TRPV4 expression in the mammalian kidney.
Functional Anatomy of TRPV4 in the Mammalian Kidney
Four reports of the cloning of TRPV4 independently noted expression of the channel in the kidney [5–8], particularly in the distal convoluted tubule. In a person of average size, the kidneys convert ~150 liters of daily glomerular filtrate (potential urine or “proto-urine”) into 0.5–1.5 liters of urine; in so doing, they also reabsorb nearly all of the filtered water and inorganic ions, as well as glucose, amino acids, and a wide variety of essential plasma constituents [54]. This 100-fold reduction in urine volume achieved along the kidney tubule is vital; in its absence, lethal volume depletion and hypotension would ensue in minutes. Such robust water conservation is achieved via a combination of apical water permeability of specific kidney tubule segments and a hyperosmotic renal medullary interstitium. Together, these factors establish both a route and a driving force for water reabsorption from the tubule lumen into the systemic circulation. Although no data unequivocally impute a role for TRPV4 in this process, clues have emerged from a detailed investigation of the distribution of TRPV4 expression in the kidney. Glomerular filtrate traverses the hyperosmotic renal medulla via the tight hairpinlike Loop of Henle (Figure 29.2A). The cells lining the “descending” arm of the loop are freely water permeant; the filtrate becomes progressively more concentrated as water traverses the tubule. An abrupt transition occurs at the genu of the loop, where the “descending” limb meets the “ascending” limb: here, apical water permeability abruptly ceases. This architecture “traps” the newly concentrated filtrate in the lumen, preventing its passive dilution by water entry as it exits the hypertonic inner medulla of the kidney. Interestingly, expression of TRPV4 is completely absent along the early parts of the kidney tubule where passive water reabsorption takes place; however, precisely at the transition between tubule water permeability and impermeability (at the hairpin turn at the base of the Loop of Henle), TRPV4 expression abruptly emerges (Figure 29.2A; [30]). TRPV4 expression continues thereafter for the length of the kidney tubule. The solitary exception is the highly specialized tubule segment called the macula densa—the only other tubule segment exhibiting constitutive apical water permeability. Here, as in the more proximal water-permeant tubule segments, TRPV4 expression is absent [30]. Therefore, TRPV4 expression in the kidney is entirely restricted to those tubule segments that lack constitutive apical water permeability and where a transcellular osmotic gradient may be expected to develop.
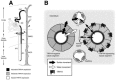
FIGURE 29.2
TRPV4 in the mammalian kidney. (A) TRPV4 expression along the mouse and rat nephron. Black shading indicates strong TRPV4 expression; gray shading indicates less intense TRPV4 expression. Throughout the course of the collecting duct, expression was stronger (more...)
A Model of TRPV4 Function in Kidney Physiology
Throughout the ascending loop and in the distal nephron, TRPV4 expression occurs predominantly at the basolateral membrane of kidney tubular epithelial cells. It is therefore unlikely that the channel comes into direct contact with the lumenal glomerular filtrate. Rather, TRPV4 is ideally situated to sense changes in osmolarity of the medullary interstitium (Figure 29.2B). Such fluctuations occur in response to a variety of physiological states and pathophysiological conditions, including even a simple change in urine flow rate. Under such conditions, TRPV4 may monitor local interstitial water balance. This balance is influenced not only by the transcellular reabsorption of solute and water from the tubular lumen into the interstitium, but also by the rate of efflux of solute and water from this tissue compartment. This latter step is accomplished by the dense capillary plexus of the cortex, and by the vasa recta penetrating the hyperosmotic kidney medulla; these envelop the kidney tubules and are charged with the herculean task of returning roughly six liters of solute-laden interstitial water to the systemic circulation each hour. Basolaterally expressed TRPV4 is well positioned to sense an imbalance between reabsorption of solute and water from the tubular lumen and efflux of these plasma constituents from the medulla via its draining capillaries.
The interstitium surrounding the proximal tubule cells (which lack TRPV4 expression) is functionally continuous with the interstitium surrounding the TRPV4-expressing cells of the cortical distal nephron; therefore, the absence of TRPV4 in the proximal nephron may serve to insulate this high-capacity reabsorptive system from responding to subtle interstitial cues. The distal nephron, in contrast, responsible for “fine-tuning” salt and water balance, may be better poised to respond to these cues. This architecture, where TRPV4-negative tubule segments accompany TRPV4-expressing segments, exists at all levels of the kidney from the cortex to the inner medulla (Figure 29.2A). For example, an acute change in osmolarity of the outer medullary interstitium could regulate activity of basolaterally expressed TRPV4 in cells of the distal nephron; the resulting intracellular calcium signal could then influence lumenal or basolateral ion transport in that tubule segment. Cells of the proximal tubule, lacking TRPV4 expression, could not respond to this signal from the interstitium. In the outer medulla, one such signal could be overwhelming of the reabsorptive capacity of the vasa recta, leading to accumulation of excess water or solute in the medullary interstitium; this stimulus in turn would be sensed by TRPV4 on the basolateral side of epithelial cells of the thick ascending limb (Figure 29.2B) and thereby “feed forward” to influence distal solute transport.
Few studies have addressed regulation of TRPV4 abundance or function in the kidney in vivo. Aminoglycoside antibiotics are widely prescribed; unfortunately, drugs of this class are limited by their potential oto- and nephrotoxicity. Kitahara and colleagues noted that administration of kanamycin, a prototypical aminoglycoside, decreased renal expression of TRPV4 [55]. This effect, however, was blunted by the antioxidant dihydroxybenzoate when it was coadministered with the antibiotic. These data are consistent with the ability of antioxidant compounds to protect the inner ear from aminoglycoside-induced cochlear hair cell damage (reviewed in reference 56). Aminoglycoside nephrotoxicity is unusual among the toxic nephropathies in that copious urinary losses of water and solute (polyuria) generally ensues [57]. Whether this clinical phenomenon is related to the downregulation of TRPV4 expression in the distal nephron—either as cause or consequence—remains to be explored.
The Role of TRPV4 in Regulating Water Balance in vivo
Sodium Balance versus Water Balance
As a putative sensor of tonicity, it is likely that TRPV4 responds to perturbations in systemic water balance rather than systemic sodium balance. Serum sodium concentration is largely independent of total body sodium balance; clinically, these two parameters are readily separable. In humans, the differential diagnosis of hypernatremia (a surrogate for plasma hypertonicity) requires an assessment of total body sodium content and water content. In nearly all circumstances, hypernatremia reflects water loss with or without sodium loss, although in rare instances it may follow ingestion or administration of concentrated sodium-containing salts (reviewed in references 58 and 59). Total body sodium balance cannot be assessed quantitatively in humans in clinical practice; rather, it is inferred from a physical examination. Findings consistent with hypovolemia (a deficit in total body sodium content) include increased heart rate and decreased blood pressure. In addition, urinary sodium excretion is quantified as a crude estimate. That is, abnormally low urinary sodium excretion is a marker for decreased total body sodium content (hypovolemia), whereas a “normal” degree of urinary sodium excretion is consistent with intravascular volume repletion and a physiological level of total body sodium content.
Classification of Hypernatremia
Despite the total body sodium content of the individual, hypernatremia nearly always reflects water loss—water loss in the absence of sodium loss in the “euvolemic” hypernatremias and water loss in excess of ongoing sodium loss in the “hypovolemic” hypernatremias. The presence or absence of associated sodium loss, while not impacting the hypernatremia per se, assists in identifying the pathological abnormality [58,59]. Euvolemic hypernatremia is caused primarily by excess urinary water loss (diabetes insipidus) or hypodipsia (decreased thirst). Nonurinary sites of water loss are also possible (e.g., water loss through the gastrointestinal tract or through perspiration). Increased water loss alone, unless overwhelming, rarely produces significant hypernatremia because it is limited by AVP action on the kidney-collecting duct (when urinary concentrating ability is preserved), and because it produces a powerful urge to drink. In the absence of an appropriate thirst response or when water is unavailable, hypernatremia can quickly ensue despite maximal urinary water conservation. Correction of euvolemic hypernatremia requires water alone. Hypovolemic hypernatremia, in contrast, results from loss of more than just pure water; “wasting” of sodium in the urine or from other sites (e.g., gut, sweat glands, etc.) is also required. If the sodium loss occurs from a nonrenal site, urinary sodium conservation is seen. Aldosterone, upregulated by the hypovolemia, minimizes urinary sodium excretion. In contrast, if the kidneys are the culprit in the sodium loss, this adaptive response is conspicuously absent. Correction of hypovolemic hypernatremia requires repletion of both water and sodium (albeit more of the former).
Water Balance in TRPV4−/− Mouse Models
It is helpful to interpret the available data for each of the two independently reported TRPV4 knockout models in light of this clinical paradigm. It is also important to emphasize, as had previously been noted, that some discrepancies between these authors’ findings might have arisen because of the different ways in which the models were generated [14].
Mizuno et al. [60] compared plasma sodium concentration in TRPV4−/− and wild-type mice. In their relatively small sample (n = 10 per group), they were unable to demonstrate a difference between the mice under basal conditions, although there was a nonsignificant trend toward higher plasma sodium concentration in the TRPV4−/− mice (140.7 + 0.8 vs. 137.9 + 1.0 mEq/l). This group also reported lower urinary sodium levels for the TRPV4−/− mice (123 + 15 vs. 165 + 21 mEq/l). In addition, they noted an insignificant but seemingly substantial decrease in systolic blood pressure among the knockout mice (105 + 4 vs. 116 + 5 mmHg). These two latter parameters are perhaps consistent with a modest relative decrease in total body sodium content and effective intravascular volume (hypovolemia); however, because these were not balance studies, the discrepancy in urinary sodium concentration might reflect in part the greater dilution of the −/− urine (see below). Therefore, no firm conclusion can be drawn about the total body sodium (“volume”) status in these mice. With respect to water balance in this study, there was a trend toward higher water loss (urine volumes of 1.8 + 0.5 vs. 1.4 + 0.4 ml/d) and greater water intake (4.5 + 1.0 vs. 4.0 + 0.8 ml/d) among the TRPV4−/− mice, although this did not reach the threshold for statistical significance. Urine volume is not a surrogate for urinary water excretion, of course, as the urine may be solute rich or solute poor, and the latter will contribute more to free water clearance. Nonetheless, consistent with these findings was the relative (and again insignificant) decrease in urinary creatinine, an indirect index of the degree of urinary concentration. With elevated plasma osmolarity, this constellation of findings could represent either (1) excess water diuresis driving thirst or (2) osmotic diuresis (e.g., from glycosuria) driving thirst. Discrimination between these scenarios requires an assessment of urine osmolarity; a relatively diluted urine is consistent with the former, whereas an “isosthenuric” urine (i.e., urine with a relatively fixed osmolarity approximating that of plasma) suggests the latter. However, the “basal” urine osmolarities were identical in the +/+ and −/−mice in this model.
Unregulated renal water loss (diabetes insipidus), which was likely a factor in the Mizuno model (Figure 29.3), generally reflects either a defect in hypothalamic production of the water-retentive peptide arginine vasopressin (AVP) or a defect in the renal response to this hormone. Perhaps consistent with the former, basal levels of serum AVP were slightly (albeit insignificantly) lower in the −/− mice. Consistent with a nephrogenic picture, however, AVP levels in response to an acute osmotic load were dramatically higher in the −/− mice relative to wild-type mice. The osmotic stimulus in this exercise was supraphysiological; the serum osmolarity was acutely increased from 310 to 410 using propylene glycol infusion. But in vitro exposure of brain slices to much more subtle hyperosmotic stimuli resulted in a similarly enhanced release of AVP from TRPV4−/− slices relative to slices from the +/+ brains. In addition, independent of AVP, excessive water diuresis can also be seen with gross abnormalities in plasma calcium or potassium concentrations, but neither were present in this model. In aggregate, the data seem most consistent with diabetes insipidus in the TRPV4−/− mice in the model of Mizuno et al. (Figure 29.3). Of note, this interpretation is based primarily on nonsignificant data trends; nonetheless, these “trends” are internally consistent and hence credible.
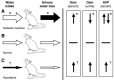
FIGURE 29.3
Genesis of a water deficit in mammals. In diabetes insipidus (A), excess urinary water loss (filled arrow) relative to normal (B) is the primary disorder; in primary hypodipsia (C), decreased water intake is primary. The thickness of the horizontal arrows (more...)
In the model of Liedtke and Friedman, plasma osmolarity was measured rather than plasma sodium concentration [61]. In contrast to the study of Mizuno et al., Liedtke found a clear-cut increase in plasma osmolarity among the TRPV4−/− mice (300 vs. 295 mosmol/kgH2O). To “unmask” this difference, however, mice were single-housed and deprived of food for 24 hours prior to assessment; in the absence of these refinements, no difference was observed. Whereas assessment of plasma osmolarity rather than plasma sodium appears to be a more direct measurement of this physiological parameter, it is subject to confounding in practice. Specifically, osmotically “ineffective” substances in the plasma such as urea (i.e., blood urea nitrogen) can contribute to measured osmolarity. However, because urea readily crosses most cell membranes, it is not considered an “effective” osmole in vivo. In contrast, other nonsodium solutes are osmotically effective in vivo (e.g., glucose) and do contribute to plasma osmolarity. Therefore, from a clinical perspective, plasma sodium concentration is frequently a more reliable index of a relative water deficit or excess than is plasma osmolarity; when the latter is used in the evaluation of human disease, simultaneous determination of blood urea nitrogen and glucose is generally obtained. Also of note in this study, mice were deprived of food but access to water was not restricted. Dietary intake is the major source of urinary urea, which contributes osmoles to the urine and can act as an osmotic diuretic. Protein intake also dictates blood urea nitrogen, which, although osmotically ineffective, influences measured osmolarity of the plasma. Therefore, protein starvation can significantly impact plasma and urine osmolarity and urine volume independently of water balance. It is unclear if these may have been factors in their study.
With respect to an assessment of volume status (total body sodium content) in the study of Liedtke and Friedman, blood pressure and urinary sodium content were not reported. It is in the realm of water balance where the striking findings emerge. In concert with an elevated plasma osmolarity, and in marked contrast to the observations of Mizuno et al., Liedtke noted dramatically reduced (~30 percent less) water intake among the TRPV4−/− mice [61]. When water was restricted for 24 hours, the discrepancy in plasma osmolarity was magnified, increasing by a further 6 percent in the −/− mice compared to only 3 percent in wild-type mice. This suggested accelerated water loss in the TRPV4 knockout mice, although data comparing urine volume or osmolarity were not available. In aggregate, these data seem most consistent with hypodipsia (i.e., a primary decrease in thirst or osmosensation; Figure 29.3) and, potentially, an inappropriate renal response to this disorder.
The data of Mizuno and colleagues [60] and of Liedtke and Friedman [61] suggest dissimilar disorders of water balance. In general, when evaluating a state of plasma hypertonicity, maneuvers designed to impact water access or water loss are paramount. Water restriction, with serial monitoring of an increment in plasma sodium (and osmolarity); detailed analysis of urine volume, osmolarity, sodium, and potassium content (i.e., in metabolic cages); and assessment of water intake would be optimal. The impact of supplemental AVP, if any, on these parameters could then be assessed. It is possible that TRPV4−/− mice will exhibit partial abnormalities at multiple levels. Tissue-specific targeting of a TRPV4 knockout to either the distal nephron or the lamina terminalis might permit dissection of these events. In the early and ground-breaking studies of these two groups of investigators, it could not be envisioned beforehand that hypertonicity would ensue in the TRPV4−/− mice; in vitro data with TRPV4 as a sensor of hypotonic stress suggested that genetic absence of this protein might predispose to water excess rather than water loss. But now the foundation has been laid for a detailed investigation into the role of TRPV4 in the regulation of systemic water balance under basal and pathological conditions.
SUMMARY
TRPV4 is activated by a variety of fatty acid metabolites and by a number of physical stimuli including thermal and osmotic stresses. It is likely that tonicity-dependent activation of TRPV4 requires SRC family kinase–dependent tyrosine phosphorylation. Other data support a role for phospholipase A2 and cytochrome P450 epoxygenase activity and involvement of epoxyeicosatrienoic acids. The architecture of subcellular localization of TRPV4 in the mammalian kidney suggests a role for this channel in responding to interstitial signals related to systemic water balance. The TRPV4-expressing tubule segments may then integrate and respond to these signals by altering lumenal solute reabsorption in a “feed forward” regulatory mechanism. The two TRPV4−/− mouse models generated to date have given rise to conflicting data: genetic absence of this sensor of hypotonicity induces a state akin to diabetes insipidus in one model and primary hypodipsia in the other. It is likely that more detailed water (and solute) balance studies will establish whether these models are as dissimilar as they initially appear, and will clarify the role of TRPV4 in regulating systemic water balance.
Acknowledgments
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health, the Department of Veterans Affairs, and the American Heart Association.
REFERENCES
- 1.
- Colbert HA, Bargmann CI. Odorant-specific adaptation pathways generate olfactory plasticity in C. elegans. Neuron. 1995;14:803–812. [PubMed: 7718242]
- 2.
- Colbert HA, Smith TL, Bargmann CI. OSM-9, a novel protein with structural similarity to channels, is required for olfaction, mechanosensation, and olfactory adaptation in Caenorhabditis elegans. J Neurosci. 1997;17:8259–8269. [PMC free article: PMC6573730] [PubMed: 9334401]
- 3.
- Minke B. Drosophila mutant with a transducer defect. Biophys Struct Mech. 1977;3:59–64. [PubMed: 870103]
- 4.
- Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2:1313–1323. [PubMed: 2516726]
- 5.
- Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103:525–535. [PMC free article: PMC2211528] [PubMed: 11081638]
- 6.
- Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2000;2:695–702. [PubMed: 11025659]
- 7.
- Wissenbach U, Bodding M, Freichel M, Flockerzi V. Trp12, a novel Trp-related protein from kidney. FEBS Lett. 2000;485:127–134. [PubMed: 11094154]
- 8.
- Delany NS, Hurle M, Facer P, Alnadaf T, Plumpton C, Kinghorn I, See CG, Costigan M, Anand P, Woolf CJ, Crowther D, Sanseau P, Tate SN. Identification and characterization of a novel human vanilloid receptor-like protein, VRL-2. Physiol Genomics. 2001;4:165–174. [PubMed: 11160995]
- 9.
- Montell C, Birnbaumer L, Flockerzi V, Bindels RJ, Bruford EA, Caterina MJ, Clapham DE, Harteneck C, Heller S, Julius D, Kojima I, Mori Y, Penner R, Prawitt D, Scharenberg AM, Schultz G, Shimizu N, Zhu MX. A unified nomenclature for the superfamily of TRP cation channels. Mol Cell. 2002;9:229–231. [PubMed: 11864597]
- 10.
- Chung MK, Lee H, Caterina MJ. Warm temperatures activate TRPV4 in mouse 308 keratinocytes. J Biol Chem. 2003;278:32037–32046. [PubMed: 12783886]
- 11.
- Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nilius B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem. 2002;277:47044–47051. [PubMed: 12354759]
- 12.
- Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M. Heat-evoked activation of the ion channel, TRPV4. J Neurosci. 2002;22:6408–6414. [PMC free article: PMC6758176] [PubMed: 12151520]
- 13.
- Gao X, Wu L, O’Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C–dependent and –independent pathways. J Biol Chem. 2003;278:27129–27137. [PubMed: 12738791]
- 14.
- Todaka H, Taniguchi J, Satoh J, Mizuno A, Suzuki M. Warm temperature-sensitive transient receptor potential vanilloid 4 (TRPV4) plays an essential role in thermal hyperalgesia. J Biol Chem. 2004;279:35133–35138. [PubMed: 15187078]
- 15.
- Lee H, Iida T, Mizuno A, Suzuki M, Caterina MJ. Altered thermal selection behavior in mice lacking transient receptor potential vanilloid 4. J Neurosci. 2005;25:1304–1310. [PMC free article: PMC6725965] [PubMed: 15689568]
- 16.
- Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, Vriens J, Cairns W, Wissenbach U, Prenen J, Flockerzi V, Droogmans G, Benham CD, Nilius B. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002;277:13569–13577. [PubMed: 11827975]
- 17.
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. [PubMed: 12879072]
- 18.
- Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol. 2004;286:C195–205. [PubMed: 14707014]
- 19.
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. [PubMed: 14595399]
- 20.
- Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–36062. [PubMed: 11451964]
- 21.
- Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004;304:265–270. [PubMed: 15031437]
- 22.
- Hilgemann DW. Biochemistry. Oily barbarians breach ion channel gates. Science. 2004;304:223–224. [PubMed: 15031439]
- 23.
- Kahn-Kirby AH, Dantzker JL, Apicella AJ, Schafer WR, Browse J, Bargmann CI, Watts JL. Specific polyunsaturated fatty acids drive TRPV-dependent sensory signaling in vivo. Cell. 2004;119:889–900. [PubMed: 15607983]
- 24.
- Kung C. A possible unifying principle for mechanosensation. Nature. 2005;436:647–654. [PubMed: 16079835]
- 25.
- Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci USA. 2004;101:396–401. [PMC free article: PMC314196] [PubMed: 14691263]
- 26.
- Xu H, Zhao H, Tian W, Yoshida K, Roullet JB, Cohen DM. Regulation of a transient receptor potential (TRP) channel by tyrosine phosphorylation. SRC family kinase–dependent tyrosine phosphorylation of TRPV4 on TYR-253 mediates its response to hypotonic stress. J Biol Chem. 2003;278:11520–11527. [PubMed: 12538589]
- 27.
- Suzuki M, Mizuno A, Kodaira K, Imai M. Impaired pressure sensation in mice lacking TRPV4. J Biol Chem. 2003;278:22664–22668. [PubMed: 12692122]
- 28.
- Praetorius HA, Spring KR. The renal cell primary cilium functions as a flow sensor. Curr Opin Nephrol Hypertens. 2003;12:517–520. [PubMed: 12920399]
- 29.
- Delmas P. Polycystins: from mechanosensation to gene regulation. Cell. 2004;118:145–148. [PubMed: 15260985]
- 30.
- Tian W, Salanova M, Xu H, Lindsley JN, Oyama TT, Anderson S, Bachmann S, Cohen DM. Renal expression of osmotically responsive cation channel TRPV4 is restricted to water-impermeant nephron segments. Am J Physiol Renal Physiol. 2004;287:F17–24. [PubMed: 15026302]
- 31.
- Teilmann SC, Byskov AG, Pedersen PA, Wheatley DN, Pazour GJ, Christensen ST. Localization of transient receptor potential ion channels in primary and motile cilia of the female murine reproductive organs. Mol Reprod Dev. 2005;71:444–452. [PubMed: 15858826]
- 32.
- Cohen D. TRPV4 and the mammalian kidney. Pflügers Archiv. 2005 [PubMed: 15988590]
- 33.
- Edashige K, Watanabe Y, Sato EF, Takehara Y, Utsumi K. Reversible priming and protein-tyrosyl phosphorylation in human peripheral neutrophils under hypotonic conditions. Archives of Biochemistry & Biophysics. 1993;302:343–347. [PubMed: 7683858]
- 34.
- Tilly BC, van den Berghe N, Tertoolen LG, Edixhoven MJ, de Jonge HR. Protein tyrosine phosphorylation is involved in osmoregulation of ionic conductances. Journal of Biological Chemistry. 1993;268:19919–19922. [PubMed: 7690749]
- 35.
- Sadoshima J, Qiu ZH, Morgan JP, Izumo S. Tyrosine kinase activation is an immediate and essential step in hypotonic cell swelling–induced ERK activation and c-fos gene expression in cardiac myocytes. Embo J. 1996;15:5535–5546. [PMC free article: PMC452298] [PubMed: 8896447]
- 36.
- Sachs JR, Martin DW. The role of ATP in swelling-stimulated K-Cl cotransport in human red cell ghosts. Phosphorylation-dephosphorylation events are not in the signal transduction pathway. J Gen Physiol. 1993;102:551–573. [PMC free article: PMC2229154] [PubMed: 8245823]
- 37.
- Good DW. Hyperosmolality inhibits bicarbonate absorption in rat medullary thick ascending limb via a protein-tyrosine kinase–dependent pathway. Journal of Biological Chemistry. 1995;270:9883–9889. [PubMed: 7730371]
- 38.
- Cohen DM. SRC family kinases in cell volume regulation. Am J Physiol Cell Physiol. 2005;288:C483–493. [PubMed: 15692147]
- 39.
- Ko BC, Lam AK, Kapus A, Fan L, Chung SK, Chung SS. Fyn and p38 signaling are both required for maximal hypertonic activation of the osmotic response element-binding protein/tonicity-responsive enhancer-binding protein (OREBP/TonEBP). J Biol Chem. 2002;277:46085–46092. [PubMed: 12359721]
- 40.
- Jiang X, Newell EW, Schlichter LC. Regulation of a TRPM7-like current in rat brain microglia. J Biol Chem. 2003;278:42867–42876. [PubMed: 12904301]
- 41.
- Vazquez G, Wedel BJ, Kawasaki BT, Bird GS, Putney JW Jr. Obligatory role of Src kinase in the signaling mechanism for TRPC3 cation channels. J Biol Chem. 2004 [PubMed: 15271991]
- 42.
- Jin X, Morsy N, Winston J, Pasricha PJ, Garrett K, Akbarali HI. Modulation of TRPV1 by nonreceptor tyrosine kinase, c-Src kinase. Am J Physiol Cell Physiol. 2004;287:C558–563. [PubMed: 15084474]
- 43.
- Hisatsune C, Kuroda Y, Nakamura K, Inoue T, Nakamura T, Michikawa T, Mizutani A, Mikoshiba K. Regulation of TRPC6 channel activity by tyrosine phosphorylation. J Biol Chem. 2004;279:18887–18894. [PubMed: 14761972]
- 44.
- Odell AF, Scott JL, Van Helden DF. Epidermal growth factor induces tyrosine phosphorylation, membrane insertion, and activation of transient receptor potential channel 4. J Biol Chem. 2005;280:37974–37987. [PubMed: 16144838]
- 45.
- Thoroed SM, Lauritzen L, Lambert IH, Hansen HS, Hoffmann EK. Cell swelling activates phospholipase A2 in Ehrlich ascites tumor cells. J Membr Biol. 1997;160:47–58. [PubMed: 9351891]
- 46.
- Basavappa S, Pedersen SF, Jorgensen NK, Ellory JC, Hoffmann EK. Swelling-induced arachidonic acid release via the 85-kDa cPLA2 in human neuroblastoma cells. J Neurophysiol. 1998;79:1441–1449. [PubMed: 9497423]
- 47.
- Pedersen S, Lambert IH, Thoroed SM, Hoffmann EK. Hypotonic cell swelling induces translocation of the alpha isoform of cytosolic phospholipase A2 but not the gamma isoform in Ehrlich ascites tumor cells. Eur J Biochem. 2000;267:5531–5539. [PubMed: 10951212]
- 48.
- Andrade YN, Fernandes J, Vazquez E, Fernandez-Fernandez JM, Arniges M, Sanchez TM, Villalon M, Valverde MA. TRPV4 channel is involved in the coupling of fluid viscosity changes to epithelial ciliary activity. J Cell Biol. 2005;168:869–874. [PMC free article: PMC2171792] [PubMed: 15753126]
- 49.
- Lang F, Busch GL, Volkl H. The diversity of volume regulatory mechanisms. Cell Physiol Biochem. 1998;8:1–45. [PubMed: 9547017]
- 50.
- Wehner F, Olsen H, Tinel H, Kinne-Saffran E, Kinne RK. Cell volume regulation: osmolytes, osmolyte transport, and signal transduction. Rev Physiol Biochem Pharmacol. 2003;148:1–80. [PubMed: 12687402]
- 51.
- Fernandez-Fernandez JM, Nobles M, Currid A, Vazquez E, Valverde MA. Maxi K+ channel mediates regulatory volume decrease response in a human bronchial epithelial cell line. Am J Physiol Cell Physiol. 2002;283:C1705–1714. [PubMed: 12388065]
- 52.
- Arniges M, Vazquez E, Fernandez-Fernandez JM, Valverde MA. Swelling-activated Ca2+ entry via TRPV4 channel is defective in cystic fibrosis airway epithelia. J Biol Chem. 2004;279:54062–54068. [PubMed: 15489228]
- 53.
- Becker D, Blase C, Bereiter-Hahn J, Jendrach M. TRPV4 exhibits a functional role in cell-volume regulation. J Cell Sci. 2005;118:2435–2440. [PubMed: 15923656]
- 54.
- Cohen D. TRPV4 and hypotonic stress. In: Wang D, editor. Molecular sensors for cardiovascular homeostasis. New York: Kluwer Academic Publishers; 2005.
- 55.
- Kitahara T, Li HS, Balaban CD. Changes in transient receptor potential cation channel superfamily V (TRPV) mRNA expression in the mouse inner ear ganglia after kanamycin challenge. Hear Res. 2005;201:132–144. [PubMed: 15721568]
- 56.
- Wu WJ, Sha SH, Schacht J. Recent advances in understanding aminoglycoside oto-toxicity and its prevention. Audiol Neurootol. 2002;7:171–174. [PubMed: 12053140]
- 57.
- Woolfson R, Hillman K. Causes of acute renal failure. In: Johnson R, Feehally J, editors. Comprehensive clinical nephrology. London: Mosby; 2000.
- 58.
- Adrogue HJ, Madias NE. Hypernatremia. N Engl J Med. 2000;342:1493–1499. [PubMed: 10816188]
- 59.
- Berl T, Kumar S. Disorders of water metabolism. In: Johnson R, Feehally J, editors. Comprehensive clinical nephrology. London: Mosby; 2000.
- 60.
- Mizuno A, Matsumoto N, Imai M, Suzuki M. Impaired osmotic sensation in mice lacking TRPV4. Am J Physiol Cell Physiol. 2003;285:C96–101. [PubMed: 12777254]
- 61.
- Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA. 2003;100:13698–13703. [PMC free article: PMC263876] [PubMed: 14581612]
- Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans.[Proc Natl Acad Sci U S A. 2003]Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans.Liedtke W, Tobin DM, Bargmann CI, Friedman JM. Proc Natl Acad Sci U S A. 2003 Nov 25; 100 Suppl 2(Suppl 2):14531-6. Epub 2003 Oct 27.
- Review TRPV Channels’ Function in Osmo- and Mechanotransduction.[TRP Ion Channel Function in Se...]Review TRPV Channels’ Function in Osmo- and Mechanotransduction.Liedtke WB. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. 2007
- Review Transient receptor potential vanilloid channels functioning in transduction of osmotic stimuli.[J Endocrinol. 2006]Review Transient receptor potential vanilloid channels functioning in transduction of osmotic stimuli.Liedtke W. J Endocrinol. 2006 Dec; 191(3):515-23.
- Review Molecular mechanisms of TRPV4-mediated neural signaling.[Ann N Y Acad Sci. 2008]Review Molecular mechanisms of TRPV4-mediated neural signaling.Liedtke W. Ann N Y Acad Sci. 2008 Nov; 1144:42-52.
- Review TRPV4: A Multifunctional Nonselective Cation Channel with Complex Regulation.[TRP Ion Channel Function in Se...]Review TRPV4: A Multifunctional Nonselective Cation Channel with Complex Regulation.Plant TD, Strotmann R. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. 2007
- The Role of TRPV4 in the Kidney - TRP Ion Channel Function in Sensory Transducti...The Role of TRPV4 in the Kidney - TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades
Your browsing activity is empty.
Activity recording is turned off.
See more...