Summary
Clinical characteristics.
MECR-related neurologic disorder is characterized by a progressive childhood-onset movement disorder and optic atrophy; intellect is often – but not always – preserved. The movement disorder typically presents between ages one and 6.5 years and is mainly dystonia that can be accompanied by chorea and/or ataxia. Over time some affected individuals require assistive devices for mobility. Speech fluency and intelligibility are progressively impaired due to dysarthria. Optic atrophy typically develops between ages four and 12 years and manifests as reduced visual acuity, which can include functional blindness (also known as legal blindness) in adulthood. Because only 13 affected individuals are known to the authors, and because nearly half of them were diagnosed retrospectively as adults, the natural history of disease progression and other aspects of the phenotype have not yet been completely defined.
Diagnosis/testing.
The diagnosis of MECR-related neurologic disorder is established in a proband with a childhood-onset movement disorder and biallelic (compound heterozygous or homozygous) pathogenic variants in MECR identified by molecular genetic testing.
Management.
Treatment of manifestations: Visual aids for decreased visual acuity due to optic atrophy; occupational therapy and physical therapy to maintain range of movement and special aids (e.g., braces, walkers, wheelchairs) to maintain/improve mobility; speech therapy for dysarthria and augmentative communication if needed. Medications that may relieve dystonia include anticholinergic agents, baclofen, and benzodiazepines.
Surveillance: The following yearly examinations are warranted: ophthalmologic (need for additional visual aids), neurologic (need for medications to relieve dystonia), speech therapy (need for augmentative communication), cognitive evaluation, and feeding evaluation (assess risk of aspiration).
Agents/circumstances to avoid: Stress and febrile illness as much as possible as these are presumed to exacerbate disease progression. Discuss anesthetic risks with a patient's medical team prior to surgical procedures.
Genetic counseling.
MECR-related neurologic disorder is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the MECR pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic diagnosis are possible.
Diagnosis
To date no formal diagnostic criteria have been published for MECR-related neurologic disorder.
Suggestive Findings
MECR-related neurologic disorder should be suspected in individuals with the following clinical findings, neuroimaging findings, and ethnicity.
Clinical findings
- Childhood-onset dystonia, chorea, and other movement disorders: ages 1-6.5 years
- Childhood-onset optic atrophy: typically ages 4-12 years. Note that optic atrophy is not necessary to consider the diagnosis of MECR-related neurologic disorder in a young child as it may appear several years after the onset of the movement disorder.
Neuroimaging findings. On MRI, bilateral hyperintense T2-weighted signal in one or more structures of the basal ganglia (i.e., caudate, putamen, or pallidum) evident at time of onset of dystonia (Figure 1)
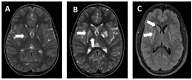
Ethnicity. Ashkenazi Jewish heritage. Note that while MECR-related neurologic disorder is more frequent in Ashkenazi Jews, it also occurs in persons of other ethnicities.
Establishing the Diagnosis
The diagnosis of MECR-related neurologic disorder is established in a proband with Suggestive Findings and biallelic (compound heterozygous or homozygous) pathogenic (or likely pathogenic) variants in MECR by molecular genetic testing (Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic MECR variants of uncertain significance (or of one known MECR pathogenic variant and one MECR variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing or a multigene panel) and comprehensive genomic testing depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of MECR-related neurologic disorder is distinctive, children with findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from other childhood-onset dystonias with bilateral symmetric basal ganglia signal intensity changes are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and imaging findings suggest the diagnosis of MECR-related neurologic disorder, molecular genetic testing approaches can include single-gene testing or use of a multigene panel.
- Single-gene testing. Sequence analysis of MECR is performed first. If only one pathogenic variant is found, gene-targeted deletion/duplication analysis could be considered; however, to date no exon or whole-gene deletions have been reported.
- A dystonia multigene panel that includes MECR and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Of note, given the novelty and rarity of MECR-related neurologic disorder, many panels for dystonia may not yet include this gene. (3) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the phenotype is indistinguishable from many other disorders of childhood-onset dystonia, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis. Note: To date such variants have not been identified as a cause of MECR-related neurologic disorder.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in MECR-Related Neurologic Disorder
Clinical Characteristics
Clinical Description
To date, the authors know of 13 individuals with MECR-related neurologic disorder: seven (5 probands and 2 family members) described by Heimer et al [2016], and six ascertained more recently by the authors (1 individual and 2 sets of sibs). The common clinical phenotype in these 13 individuals is characterized by childhood-onset movement disorder followed by optic atrophy, and often – but not always – preserved intellect. Similar to other metabolic disorders, symptoms may fluctuate temporally with febrile illnesses. In one instance, the young child never fully regained motor skills lost after fever. Because of the limited number of affected individuals reported to date, and because nearly half of them were diagnosed retrospectively as adults, the natural history of progression of the known features of the disorder as well as other possible aspects of the phenotype have not yet been completely defined.
The motor disability and dysarthria progress with time. Severity may vary between affected individuals, even within the same family. Heimer et al [2016] describe a male age 46 years (Family C, Patient II:2) with unintelligible speech and contractures who was confined to a wheelchair and totally dependent for all activities of daily living, whereas his brother age 28 years (Patient II:8) walked independently despite limb dystonia and had slightly slurred dysarthric – but intelligible – speech.
In a different family of two affected brothers (ages 5 and 6 years), one had progressive dystonia, spasticity, and ataxia whereas the other had predominantly hypotonia and neck muscle weakness; neither has walked independently [Family F; Author, unpublished observations].
In a third family with three affected sisters, the oldest (currently age 15 years) started walking at about age two years, then exhibited pronounced chorea from age 5.5 years and dystonia from age 12 years. The youngest (currently age 4 years) exhibited arm dystonia at age one year, walked independently at age 22 months, and currently manifests dystonia of all limbs and facial chorea. In contrast, the middle sister (currently age 5.5 years) started walking at age 12 months; neurologic examination revealed minimal dystonia of the left leg of which the parents had previously been unaware [Family G; Author, unpublished observations].
Neurologic manifestations. The presenting manifestation is an involuntary movement disorder, mainly dystonia that can be accompanied by chorea and/or ataxia.
Onset of the movement disorder is during early childhood (12 months–6.5 years). However, a history of hypotonia, increased laxity, and delayed motor development from the first year of life is possible.
The motor disability gradually progresses; over time, some affected individuals require a walker or wheelchair for ambulation. Some with earlier onset and more rapid progression may never walk independently.
Speech fluency and intelligibility are progressively impaired due to dysarthria. In some cases (e.g., the three sisters in Family G) articulation may be impaired at onset of speech, whereas in other children (e.g., the two brothers in Family F) speech may never develop despite relative preservation of receptive language.
Cognition was unaffected or relatively spared in five of the seven reported by Heimer et al [2016] (Family E). Of two brothers with impaired cognition, linguistic skills and executive function deteriorated to an extremely low range at age nine years in one (Patient II:1), whereas the other (Patient II:3) was reported to have low average verbal comprehension with extremely low function on the other WISC IV indices.
Although the oldest of the three sisters in Family G was suspected of having polyneuropathy (areflexia and decreased sensation noted around age two years), these findings were not evident on more recent examination. Furthermore, conflicting results of two nerve conduction velocity (NCV) tests several years apart cast doubt on whether these findings resulted from polyneuropathy or were other manifestations of MECR-related neurologic disorder.
To date, seizures and encephalopathy have not been reported.
Ocular manifestations. Optic atrophy develops within seven years of the onset of dystonia (i.e., between ages 4 and 12 years). It manifests as reduced visual acuity, which can include functional blindness in adulthood in some individuals.
Abnormal eye movements (nystagmus or roving eye movements) can also be seen.
Life expectancy. All currently known affected individuals are alive; two are in their fifth decade.
Laboratory tests. No consistent biomarker was found. Of note, elevated urinary 3 OH-isovaleric acid was found in one individual and 3 methyl glutaconic acid in another.
Genotype-Phenotype Correlations
No clear genotype-phenotype correlations have been observed. Of the 13 currently known affected individuals, 12 are compound heterozygotes with various combinations of a missense variant and nonsense variant, and one is a homozygote for a missense variant [Heimer et al 2016 and personal communication]. These findings suggest that the combination of two nonsense variants could either be incompatible with life or give rise to a more severe phenotype.
Prevalence
To the authors' knowledge, only 13 individuals from eight families have been diagnosed with MECR-related neurologic disorder to date. Five of the eight families were of Ashkenazi Jewish origin, suggesting possible increased prevalence in this population [Heimer et al 2016 and personal communication].
Unpublished results based on the Inflammatory Bowel Disease Exomes Browser containing more than 5,500 exomes of Ashkenazi Jewish individuals revealed an increased frequency of two pathogenic variants among the Ashkenazi Jewish population:
- c.695G>A missense variant in 18/5,598; variant frequency of 1:311
- c.830+2dupT splice site variant in 41/5,576; variant frequency of 1:136
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in MECR.
Differential Diagnosis
The differential diagnosis of MECR-related neurologic disorder includes disorders that combine the clinical features of childhood-onset movement disorder (mainly dystonia [see Hereditary Dystonia Overview] but also ataxia and chorea) and the MRI findings of signal abnormality in the basal ganglia present when the movement disorder appears.
Table 2.
Disorders with Dystonia and MRI Signal Abnormality in the Basal Ganglia to Consider in the Differential Diagnosis of MECR-Related Neurologic Disorder
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with MECR-related neurologic disorder, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
The following are appropriate:
- Visual aids can be used in cases of decreased visual acuity due to optic atrophy.
- Physiotherapy can be used to maintain range of movement.
- Occupational therapy can be used as appropriate to develop and maintain skills related to activities of daily living, which will vary across the life span.
- Special aids such as braces, walkers, and wheelchairs can maintain/improve mobility.
- Speech therapy, if speech dysarthria is present, and assessment for augmentative communication devices
- Medications that may relieve dystonia, such as anticholinergic agents, baclofen, and benzodiazepines, can be considered.
- Anticholinergic agents act peripherally on the neuromuscular junction, but can have a variety of adverse central nervous system (CNS) effects.
- Baclofen, which works on GABAB receptors and functions as a CNS depressant and skeletal muscle relaxant, can be administered either through an intrathecal pump or systemically (enterally).
- Benzodiazepines, which are GABAA agonists, can reduce muscle tone and alleviate the dystonia; however, they also cause sedation.
- Deep brain stimulation (DBS): While some patients with severe dystonia are treated with DBS, experience with MECR-related neurologic disorder is limited. To date, there are no reports of individuals with this disorder treated with DBS. Moreover, due to the existence of basal ganglia lesions, DBS may not be suitable for many persons with MECR-related neurologic disorder.
- Of the three sisters (Family G; Author, unpublished observations), two are treated for ADHD: one with Vyvanse® (lisdexamfetamine dimesylate), which seems to also improve her dystonia and dysarthria; the other with Adderall, which seems to improve her dystonia and balance.
Surveillance
The following are recommended:
- Yearly eye examination to determine need for additional visual aids
- Yearly neurologic assessment to determine need for additional interventions, including speech therapy
Agents/Circumstances to Avoid
Disease progression is presumed to be exacerbated by stress or febrile illness; therefore, prevention of these – to the extent possible – is recommended.
One patient reported onset of significant new, long-term motor symptoms following extended anesthesia with propofol. As with other mitochondrial disorders, anesthetic considerations should be discussed with a patient's medical team prior to any surgical procedure [Niezgoda & Morgan 2013].
In one of the sisters (Family G), a test dose of dopamine worsened her chorea significantly.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
The pathomechanism of MECR-related neurologic disorder suggests the possible therapeutic effect of supplementation with lipoic acid (LA) and octanoic acid (C8). One individual showed remarkable improvement after receiving LA and a nutritional supplement high in C8 within three months of symptom onset [Heimer et al 2016]. The authors are currently offering their patients treatment with a Mito Cocktail of coenzyme Q10, riboflavin, thiamine, and alpha lipoic acid with the addition of octanoic acid, vitamin E, and vitamin C (see also Author Notes). Of note, the efficacy of this regimen for treatment of MECR-related neurologic disorder has yet to be proven in an evidence-based manner.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
MECR-related neurologic disorder is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one MECR pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. Unless an affected individual's reproductive partner also has MECR-related neurologic disorder or is a carrier, offspring will be obligate heterozygotes (carriers) for a pathogenic variant in MECR.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an MECR pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the MECR pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the MECR pathogenic variants have been identified in an affected family member, prenatal testing and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- United Mitochondrial Disease FoundationPhone: 888-317-UMDF (8633)Email: info@umdf.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
MECR-Related Neurologic Disorder: Genes and Databases
Table B.
OMIM Entries for MECR-Related Neurologic Disorder (View All in OMIM)
Gene structure. MECR (NM_016011.4) comprises ten exons and a total of 2,539 bp. Multiple transcript variants have been reported. (For a detailed summary of gene, transcript, and protein information, see Table A, Gene.)
Pathogenic variants. To date, six pathogenic variants have been described: three missense, two nonsense, and one splice site [Heimer et al 2016]; see Table A, Databases. The frequency of two pathogenic variants is increased in Ashkenazi Jewish populations (see Prevalence; Table 4).
Table 4.
MECR Pathogenic Variants Discussed in This GeneReview
Normal gene product. MECR encodes mitochondrial trans-2-enoyl-coenzyme A-reductase (MECR); the transcript NM_016011.4 encodes a 373-amino acid protein (NP_057095.4) with four main domains: mitochondrial transit peptide, NADPH cofactor-binding domain, and two catalytic domains. MECR catalyzes the last step of human mitochondrial fatty acid synthesis (mtFAS), turning trans-2-enoyl-ACP into acyl-ACP. MECR also serves as the precursor for lipoic acid synthesis, which functions as a cofactor for key enzymes of the respiratory chain [Hiltunen et al 2009].
Abnormal gene product. Decreased MECR activity reduces production of octanoyl-ACP (which regulates mitochondrial RNA processing and translation) and reduces respiratory complex assembly [Kursu et al 2013].
Chapter Notes
Author Notes
Dr Gali Heimer is a Senior Pediatric Neurologist and Director of the Angelman Clinic in the Pediatric Neurology Unit of the Edmond and Lily Safra Children's Hospital, and a member of the Talpiot Medical Leadership Program at the Sheba Medical Center.
Her clinical work and research focuses on neurogenetics: diagnosing known and novel genetic causes of rare neurologic diseases of childhood with emphasis on unraveling their pathomechanism and searching for therapeutic strategies.
The authors plan to test the effect of LA/C8 on cell lines from individuals with MECR-related neurologic disorder and – if favorable – to perform a clinical trial of LA/C8 supplementation. Clinicians are encouraged to contact the authors before initiating LA/C8 supplementation in an affected individual to obtain current dosing recommendations and to allow for prospective collection of clinical data pre- and post-treatment.
Revision History
- 9 May 2019 (bp) Review posted live
- 12 June 2017 (gh) Original submission
References
Literature Cited
- Heimer G, Kerätär JM, Riley LG, Balasubramaniam S, Eyal E, Pietikäinen LP, Hiltunen JK, Marek-Yagel D, Hamada J, Gregory A, Rogers C, Hogarth P, Nance MA, Shalva N, Veber A, Tzadok M, Nissenkorn A, Tonduti D, Renaldo F, Kraoua I, Panteghini C, Valletta L, Garavaglia B, Cowley MJ, Gayevskiy V, Roscioli T, Silberstein JM, Hoffmann C, Raas-Rothschild A, Tiranti V, Anikster Y, Christodoulou J, Kastaniotis AJ, Ben-Zeev B, Hayflick SJ, et al. MECR mutations cause childhood-onset dystonia and optic atrophy, a mitochondrial fatty acid synthesis disorder. Am J Hum Genet. 2016;99:1229–44. [PMC free article: PMC5142118] [PubMed: 27817865]
- Hiltunen JK, Schonauer MS, Autio KJ, Mittelmeier TM, Kastaniotis AJ, Dieckmann CL. Mitochondrial fatty acid synthesis type II: more than just fatty acids. J Biol Chem. 2009;284:9011–5. [PMC free article: PMC2666548] [PubMed: 19028688]
- Kursu VA, Pietikäinen LP, Fontanesi F, Aaltonen MJ, Suomi F, Raghavan Nair R, Schonauer MS, Dieckmann CL, Barrientos A, Hiltunen JK, Kastaniotis AJ. Defects in mitochondrial fatty acid synthesis result in failure of multiple aspects of mitochondrial biogenesis in Saccharomyces cerevisiae. Mol Microbiol. 2013;90:824–40. [PMC free article: PMC4153884] [PubMed: 24102902]
- Niezgoda J, Morgan PG. Anesthetic considerations in patients with mitochondrial defects. Paediatr Anaesth. 2013;23:785–93. [PMC free article: PMC3711963] [PubMed: 23534340]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
Publication Details
Author Information and Affiliations
Pinchas Borenstein Talpiot Medical Leadership Program
Sheba Medical Center
Tel HaShomer, Israel
Oregon Health & Science University
Portland, Oregon
Oregon Health & Science University
Portland, Oregon
Pediatrics and Neurology
Oregon Health & Science University
Portland, Oregon
Sheba Medical Center
Tel HaShomer, Israel
The Sackler School of Medicine
Tel Aviv University
Tel Aviv, Israel
Publication History
Initial Posting: May 9, 2019.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Heimer G, Gregory A, Hogarth P, et al. MECR-Related Neurologic Disorder. 2019 May 9. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
