Clinical Description
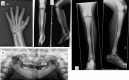
Pycnodysostosis is characterized by short stature, typical facial appearance (small jaw with obtuse mandibular angle and convex nasal ridge), osteosclerosis with increased bone fragility, acroosteolysis of the distal phalanges, delayed closure of the cranial sutures, and dysplasia of the clavicle. In affected individuals, the facial features become more prominent with age, likely due to progressive acroosteolysis of the facial bones, but can usually be appreciated from early childhood, particularly the small jaw and convex nasal ridge [Turan 2014].
A comprehensive review of previously published reports [Xue et al 2011] identified 159 individuals including 59 unrelated families with confirmed homozygous or compound heterozygous pathogenic variants in CTSK. A further 27 affected individuals from 17 unrelated families were recently described, with molecular data available for 14 families [Bizaoui et al 2019]. The following description of the phenotypic features associated with pycnodysostosis is based on these reports.
Table 2.
Pycnodysostosis: Frequency of Select Features
View in own window
| Feature | % of Persons w/Feature |
|---|
|
Clinical
| Short limb, short stature | ~100% |
| Intrauterine growth restriction | ~30% |
| Brachydactyly | >90% |
| Frontal bossing | >80% |
| Persistently open anterior fontanelle | 80% |
| Convex nasal ridge | ~70% |
| Small jaw | >70% |
| Midface retrusion | 60% |
| Proptosis | 60% |
| Blueish sclerae | 30%-40% |
| Obstructive sleep apnea | >65% |
| Increased incidence of fractures | ~70% |
| Nail anomalies | >50% |
| Dental anomalies | 30%-40% |
|
Radiographic
| Osteosclerosis | ~100% |
| Acroosteolysis of the terminal phalanges | >90% |
| Non-pneumatized mastoids | 80% |
| Delayed fusion of cranial sutures | 67% |
| Obtuse mandibular angle | 65% |
| Clavicular dysplasia | 25% |
Growth deficiency / short stature. Short stature is reported in almost 100% of individuals with pycnodysostosis. Individuals typically develop short stature by early childhood with decreased growth velocity, although 30% are reported to have intrauterine growth deficiency. Limbs are often disproportionately short compared to the trunk, with rhizo-, meso-, and acromelia. Documented adult heights are typically <150 cm for males (average 2.9 SD below the mean) and 130-134 cm for females (average 4.1 SD below the mean) [Bizaoui et al 2019].
About 50% have growth hormone deficiency but almost all have low IGF-1 levels. Administration of growth hormone has been shown to result in a satisfactory elevation in IGF-1 levels and near-normalization of adult height and skeletal proportions [Rothenbühler et al 2010].
Individuals with a growth hormone deficiency often also have pituitary hypoplasia identified on head imaging; no other abnormalities in pituitary hormones or pubertal development have been detected [Turan 2014].
Three individuals (2 diagnosed clinically and 1 with a molecular diagnosis) have been reported with taller-than-expected stature including an adult Mexican male of 153 cm (-1.9 SD), an adult Mexican female of 150 cm (-0.6 SD), and a Chinese boy age eleven years with normal height (137cm; -0.9 SD) [Zheng et al 2013, Valdes-Flores et al 2014].
Craniofacial appearance. The characteristic facial features (midface retrusion due to hypoplastic maxilla and small jaw with an obtuse mandibular angle) can become more apparent with age but are often detectable in infants, along with large anterior and posterior fontanelles and open cranial sutures with frontal and parietal bossing [Appelman-Dijkstra & Papapoulos 2016]. Additional common facial features include a convex nasal ridge. Less common features include proptosis with blueish sclera, and cleft palate or high palate with a midline groove [Bizaoui et al 2019]. The apparent palatal midline groove is due to narrow palate with shallow vault and fallen palatal wings with prominent median palatal raphe in eight individuals studied by Otaify et al [2018].
Skeletal. The second most common feature (after short stature) is increased bone density (osteosclerosis), which occurs throughout the skeleton and is progressive. The medullary canals, while often narrowed, remain present with evidence of hematopoiesis.
More than 90% of reported individuals have short hands and feet with short digits and progressive acroosteolysis of the terminal phalanges of the fingers and toes. Short metatarsals and metacarpals have not been described.
Other common imaging features include non-pneumatized mastoids (80%) and delayed fusion of the skull sutures (67%). The clavicles may be dysplastic (25%) with acroosteolysis of the acromial end. Less common features include wormian bones (18%), mild scoliosis (12%), leg length discrepancy (8%), spondylolysis, spondylolisthesis, and narrow ilia. Coronal craniosynostosis has been reported in four individuals [Bertola et al 2010, Caracas et al 2012, Bizaoui et al 2019]. Chronic pain is reported in up to 60% of adults with pycnodysostosis, with onset usually in the third decade [Bizaoui et al 2019].
Bone fragility. Individuals with pycnodysostosis have an increased fracture rate with an average 0.2 fractures per year and an average age of first fracture around age ten years [Bizaoui et al 2019]. The youngest reported individual with a fracture was age ten months; This individual had two sibs who died, reportedly from the same disorder, suggesting a more severe phenotype or genotype; however, molecular studies were not performed [Caracas et al 2012].
Fracture healing is often delayed with incomplete remodeling. Surgical fixation is often complicated by narrow medullary canals, and sclerotic bone poses an increased risk of intraoperative iatrogenic fracture [Grewal et al 2019]. To date, no effective pharmaceutical treatments have been established for the bone fragility. Bisphosphonate therapy is contraindicated due to underlying osteoclast dysfunction in pycnodysostosis.
ENT. Stridor and laryngomalacia (20%) are not uncommon manifestations, and can lead to an early suspicion of pycnodysostosis. Obstructive sleep apnea (OSA) is frequently reported (>60%), and can be particularly severe in children with pycnodysostosis. Of those with OSA, 48% required noninvasive ventilation between ages five and ten years [Testani et al 2014, Bizaoui et al 2019]. Mild conductive hearing loss occurs in up to 50% of individuals [Bizaoui et al 2019].
Dental abnormalities include delayed eruption of the deciduous and permanent teeth, persistence of deciduous teeth (resulting in a double row of teeth), hypodontia, malocclusion, enamel hypoplasia, and increased caries [Turan 2014, Khoja et al 2015, Otaify et al 2018].
Nails are often flat, grooved, and dysplastic. The skin may be wrinkled over the dorsa of the fingers, secondary to shortened digits and acroosteolysis.
Neurologic. Intelligence is typically normal in affected individuals unless a brain malformation is present. Mild psychomotor difficulties have been reported in up to 30% of individuals [Bizaoui et al 2019]. Rarely reported neurologic abnormalities include Chiari malformation (1 individual), cerebral demyelination (3 individuals), and pyramidal syndrome (1 individual) [Soliman et al 2001, Stark & Savarirayan 2009, Bizaoui et al 2019].
Ocular abnormalities have been reported, including refractive disorders and strabismus. One individual was reported to have severe vision loss as a result of intracranial hypertension and papilledema [Bizaoui et al 2019].
Obesity has not been reported as a typical feature of pycnodysostosis; however, in a cohort of 27 individuals, 26% were found to be overweight [Bizaoui et al 2019].
Prognosis. Individuals with pycnodysostosis usually have normal life expectancy.
Other. Less commonly reported features include joint laxity, deformities of the chest shape (narrow chest, kyphosis, and lordosis), and hepatosplenomegaly. An ectopic pelvic kidney and unexplained pancytopenia have each been reported in one individual.