Introduction
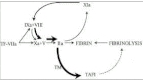
Since 1991, the opinion on the role of factor XI in coagulation has changed from an intermediary between the contact system and factor IX to an important protease in both the coagulation and the fibrinolytic systems. This change was initiated by two publications that demonstrated the capability of thrombin to activate factor XI.1,2 This activation pathway provided an explanation for an already longer existing clinical enigma. Patients deficient in factor XII, which should be the main activator of factor XI, do not suffer from a bleeding tendency, in contrast to factor XI-deficient patients who do have a mild hemorrhagic diathesis.3,4 In the revised model of coagulation, factor XI is activated by thrombin which is formed by the extrinsic pathway. This activation pathway is one of the feed-back loops by which thrombin maintains its own formation after tissue factor pathway inhibitor (TFPI) blocks the tissue factor/factor VIIa-factor Xa complex.5 Later studies have shown that this additional formed thrombin is not only important for the generation of fibrin but also for the activation of a fibrinolysis inhibitor called thrombin activatable fibrinolysis inhibitor (TAFI). Activated TAFI downregulates the fibrinolytic system resulting in protection of the fibrin clot.6 In this Chapter we will discuss the role and molecular mechanisms of factor XI and TAFI activation with special attention to disseminated intravascular coagulation (DIC).
Biochemistry of Factor XI
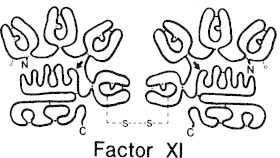
Factor XI is a dimer, consisting of two identical subunits of 80 kDa connected by a single disulfide bond (Fig. 1). The plasma concentration is 3 to 7 μg/ml (20 to 40 nM), almost all of which circulates as a complex with high molecular weight kininogen (HMWK).7 Like most clotting factors, factor XI is a zymogen that requires proteolytic activation to become an active serine protease. The proteolytic activation is achieved by cleavage at the Arg 369-Ile 370 peptidyl bond, to yield two heavy chains of 50 kDa and two light chains of 35 kDa linked by disulfide bonds.1,8

The amino acid sequence and domain structure of factor XI have been elucidated in 1986.9 Each light chain contains an active site consisting of a catalytic triad of His-413, Asp-462 and Ser-557.10 Each heavy chain consists of four repeat sequences or apple domains called A1, A2, A3, A4 and is 58% identical to the corresponding region of prekallikrein.9 The A1 domain harbors the binding site for HMWK as well as the site for interaction with (pro)thrombin.11–13 The A2 domain contains a site necessary for the Ca2+-dependent activation of factor IX by factor XIa,14,15 although recent studies have shown that the binding site for factor IX is on the A3 domain.16 The A3 domain is capable of binding platelets in the presence of HMWK,17 and also contains a binding site for unfractionated heparin.18 The binding sites on factor XI identified for the interactions with factor IX and platelets are in close proximity to each other and that raises questions about factor XI binding simultaneously to platelets and factor IX via the same A3 domain. Recently, Sun et al suggested that because factor XI is a dimer it might bind to platelets via one A3 domain and uses the other A3 domain to interact with factor IX.19 Finally, the A4 domain binds factor XII and mediates the dimer formation between the two polypeptide chains.20-22
Activation of Factor XI
Factor XI can be activated in vitro by factor XIIa, factor XIa and thrombin. During contact activation, factor XII becomes auto-activated upon binding to negatively charged surfaces where it can activate factor XI. HMWK is an important co-factor in this reaction assembling and stabilizing factor XI in a conformation facilitating its activation.23 HMWK also forms a complex with prekallikrein, and the interaction of this complex with factor XIIa results in the formation of kallikrein, which in turn releases the nonapeptide bradykinin from HMWK.4 As was demonstrated in a baboon model of lethal E. coli septic shock, inhibition of factor XIIa activity en thereby the formation of bradykinin, prevented hypotension but not DIC,24 strongly suggesting that factor XIIa is not involved in the activation of factor XI in DIC.
Thrombin-mediated factor XI activation and auto-activation of factor XI were first described on non-physiological surfaces like dextran sulfate. Thrombin can activate factor XI in the absence of co-factors or a surface (Fig. 2), but kinetic studies indicate that this reaction is too slow to be physiologically relevant.2 Moreover, fluid-phase thrombin is inhibited by antithrombin within seconds while fibrin-bound thrombin is still active but protected from the inhibitory actions of antithrombin.25,26 Therefore, the activation of factor XI by thrombin in vivo is probably a localized reaction which can take place on one of the various surfaces in the human vasculature, for example on the surface of the fibrin clot.
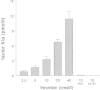
Besides dextran sulfate also more physiological polyanions like glycosaminoglycans, which are present on the vascular subendothelium, have been demonstrated to act as co-factors in thrombin mediated activation of factor XI. Heparin at a concentration of 5 μg/ml induced a 60-fold increase in activation rate, while dermatan sulphate increased a 12-fold increase in factor XI activation.27
Platelets can also serve as a physiological surface for thrombin-mediated factor XI activation. Factor XIa can bind to platelets in the presence of HMWK and retains full capacity to activate factor IX.28 Furthermore, activated platelets promote thrombin mediated factor XI activation in the absence of factor XII at rates greater than obtained with dextran sulfate.29,30
Questions were raised about factor XI activation by thrombin in a plasma-milieu since it was demonstrated that HMWK and fibrinogen can inhibit this reaction.2,31 However, these experiments were performed using high concentrations of dextran sulfate as co-factor, which is a poor choice because dextran sulfate has an antithrombin effect in plasma. Furthermore, HMWK inhibits strongly in the presence, yet only slightly on the absence of dextran sulfate.32 In the presence of thrombin, fibrin rather than fibrinogen will be present in plasma and it was demonstrated that factor XI activation by thrombin is not affected by binding of thrombin to fibrin.33
Factor XI activation in a plasma milieu has been demonstrated using gel electrophoresis, autoradiography and a detection system measuring change in turbidity as a measure of fibrin formation.34,35 Since already less than 1% activation of factor XI can result, via the amplification system of the intrinsic pathway, in significant amounts of thrombin it is easier to measure factor XI activation indirectly. Using this indirect system of measuring factor XI activation, von dem Borne et al clearly demonstrated a role of factor XI not only in fibrin formation but also in fibrinolysis.35 In a series of experiments it was shown that factor XI activation could contribute to formation of fibrin when coagulation was initiated with low concentrations of tissue factor. At higher tissue factor concentrations factor XI had no effect on the amount of fibrin formed. A key observation was that the presence of factor XI inhibited clot lysis, induced by adding tissue type plasminogen activator (t-PA), even at tissue factor concentrations at which factor XI had no effect on fibrin formation. It was demonstrated that the inhibitory effect of factor XI on fibrinolysis was caused by the generation of additional thrombin, formed after clot formation has taken place.35 So, on the one hand only small amounts of thrombin provided by the extrinsic pathway were sufficient for clot formation, whereas on the other hand large amounts of thrombin formed by the intrinsic pathway were necessary for protecting the clot against lysis.
The intermediary between factor XI-dependent thrombin formation and fibrinolysis was later elucidated as a carboxypeptidase called thrombin activatable fibrinolysis inhibitor (TAFI).
Biochemistry of TAFI
The “discovery” of TAFI followed from efforts to find an explanation for the pro-fibrinolytic capacity of the anticoagulant protein activated protein C. Bazjar et al found that activated protein C is not intrinsically profibrinolytic but that by attenuating thrombin formation a fibrin clot becomes more susceptible to fibrinolysis.36 When massive prothrombin activation occurs an antifibrinolytic activity was generated and the search for a thrombin activatable inhibitor of fibrinolysis started. The protein that was found was a zymogen, which could be activated by thrombin to an enzyme with carboxypeptidase B-like activity.37 This enzyme had been described before by other investigators and had been named plasma carboxypeptidase B or carboxypeptidase U (unstable).38,39 TAFI is a single-chain glycoprotein of about 60 kDa, synthesized by the liver, and circulates in human plasma at a concentration of 4–15 μg/ml (75 to 275 nM).40,41 TAFI antigen levels are the same in men and women.42 TAFI is activated by cleavage at Arginine 92 by thrombin and activated TAFI (TAFIa) can inhibit t-PA induced fibrinolysis.40 In normal individuals, the TAFI antigen levels correlated with TAFI activity.41 The activation of TAFI is strongly stimulated by the thrombin-thrombomodulin complex at an efficiency 1250-fold greater than by thrombin alone.43 Thrombomodulin is a component of the blood vessel wall, but also soluble fragments of thrombomodulin are present in the circulation and both forms of thrombomodulin can potentiate the activation of TAFI.44,45 Thrombomodulin binds thrombin and changes the specificity of thrombin from fibrinogen to protein C, resulting in an anticoagulant rather than a procoagulant activity. The existence of two substrates for the thrombin-thrombomodulin complex implies a dual function: anticoagulant by activating protein C and antifibrinolytic by activating TAFI. It seems that the local thrombomodulin concentration is an important factor in the regulation of these opposite effects.46 Protein C and TAFI both require epidermal growth factor (EGF) domains 4,5, and 6 of thrombomodulin for efficient activation, but TAFI activation requires approximately 13 additional residues amino acids extra in domain 3.47,48
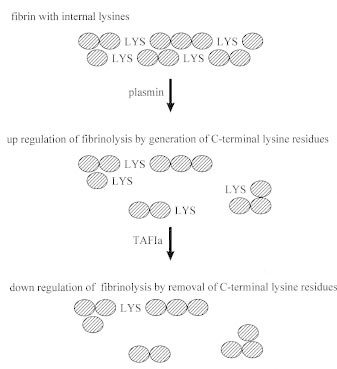
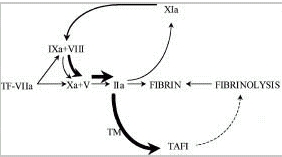
TAFIa catalyzes removal of C-terminal lysine and arginine residues from fibrin. These sites on the fibrin monomers are generated as a result of partial cleavage by plasmin and are necessary to enhance binding of plasminogen to fibrin, which facilitates fibrinolysis (Fig. 3).49 At higher concentrations TAFIa also directly inhibits plasmin.50

No natural inhibitor of TAFIa has been described, but probably its intrinsic instability especially at body temperature is important for its down regulation in vivo.51
The Intrinsic Pathway and TAFI Activation
It is important to realize that the largest production of thrombin is after initial clot formation has taken place. The fibrin clot itself provides an environment in which the enzyme thrombin is partly protected against inhibition by antithrombin.26
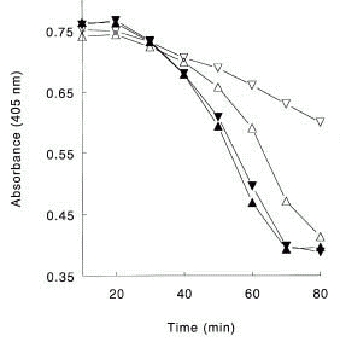
In whole blood systems and in systems working with purified components it has been demonstrated that the intrinsic pathway consisting of clotting factors XI, IX and VIII, is responsible for the major part of the total amount of thrombin formed.52,53 Deleting factor VIII or IX from a reaction system with purified components had no effect on the initial rate of thrombin observed, but the propagation rate was only one third of that observed from a system including these intrinsic factors at plasma concentrations. The addition of factor XI to this experimental system increased the rate of thrombin formation by 15%.52Using a clot lysis assay von dem Borne et al demonstrated that the mechanism of factor XI dependent inhibition of fibrinolysis is dependent upon TAFI.6 Inhibiting the activation of TAFI with a monoclonal antibody or the activity of factor XIa with a monoclonal antibody resulted in the same shortening of the lysis time. In plasma deficient of TAFI the factor XIa-inhibiting antibody had no extra effect on clot lysis (Fig. 4). This antifibrinolytic effect, which is TAFI dependent, has also been demonstrated for clotting factors IX and VIII, the other factors of the intrinsic pathway.54 Using a very sensitive functional assay for TAFIa quantification in plasma of 13 individuals it was shown that on average 65% (range 35–89%) of the total amount of TAFIa formed is factor XI dependent.55
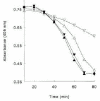
In Vivo Fibrinolysis Models
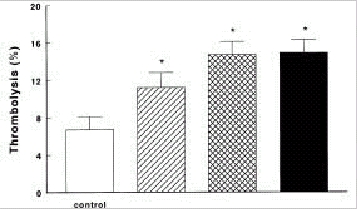
The antifibrinolytic role of activation of factor XI and TAFI has been studied in several animal models. Minnema et al have demonstrated in a rabbit thrombosis model that inhibiting factor XIa activity within the fibrin clot resulted in an almost two-fold increase in endogenous thrombolysis of jugular vein thrombi compared with controls.56 Inhibiting TAFIa activity with a potato carboxypeptidase inhibitor also resulted in a two-fold increase of fibrinolysis and combined inhibition of TAFIa activity and factor XI activation did not further increase fibrinolysis (Fig. 5). In addition, inhibiting systemic factor XIa activity enhanced endogenous thrombolysis in this model suggesting that thrombin formation continues in a factor XI dependent way after initial clot formation has taken place and that this thrombin is capable of activating TAFI.
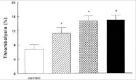
In a dog arterial thrombosis model, Redlitz et al demonstrated that during thrombolysis induced with t-PA, TAFI is activated and that there was a positive correlation with TAFIa activity and the time to restoration of blood flow in this model.57 This was further explored by Klement et al in a rabbit arterial thrombolysis model.58 The authors demonstrated in their thorough experimental set up that co-administration of potato carboxypeptidase inhibitor, as inhibitor of TAFIa, and t-PA significantly improved t-PA induced thrombolysis measured as a significant increase in original clot lysis, a decrease in the time to aortic patency and an increase in the duration of aortic patency. These results were achieved without side effects like enhanced bleeding. Inhibiting TAFIa activity may be the first clinical application in patients treated with t-PA because of acute arterial thrombosis.
In Vivo DIC Models
Only a few data reports have been published in abstract form about the role of TAFI in experimental models of DIC.59,60 In these rat models, DIC was induced after administration of endotoxin or tissue factor. TAFIa activity was inhibited with a potato carboxypeptidase inhibitor or with DL-mercaptmethyl-3-guanidinoeththylthio-propanoic acid and the lungs or kidneys were excised and examined for fibrin deposition and compared with controls. Both inhibitors of TAFIa activity reduced the amount of fibrin deposition in the lungs or kidneys. However preliminary, these data imply that also in DIC TAFI is activated and that inhibiting TAFIa can reduce the amount of fibrin deposition.
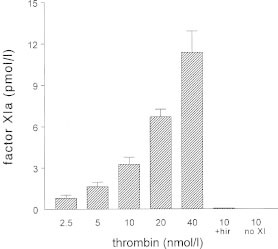
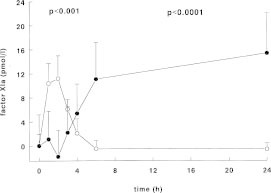
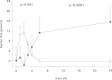
In a human experimental model of DIC factor XIa activity could be detected already one hour after a single bolus injection of endotoxin to human volunteers (Fig. 6).61 The primary set up of this study was to examine the activation pathways of factor XI in vivo. Besides measuring factor XIa activity in an enzyme capture assay, activated factor XI complexed to its inhibitors was also measured. Thrombin activity was measured as thrombin-antithrombin complexes and with the prothrombin fragment F1+2. Activation of factor XII and prekallikrein was also measured using ELISAs. Using these sensitive assays no activation of the contact system could be demonstrated, while thrombin generation was detected after one hour to become maximal after 3 to 4 hours. Activation of factor XI persisted for more than 4 hours. This activation was accompanied by a small but significant decline in factor XI antigen levels after 2 hours, which returned to baseline at 24 hours. Using a computer simulation model it was calculated that the amount of activated factor XI generated was approximately 670 pM, which corresponds with 2 to 3% of the plasma concentration of factor XI.61
Patients with Disseminated Intravascular Coagulation
Activation of factor XI has been measured in small patient groups with DIC. In one study factor XI antigen levels in 9 patients with suspected DIC were normal, while in another publication a patient with severe eclampsia and DIC had a marked reduction of factor XI clotting activity.62,63 Because only small amounts of factor XI are activated in patients it is difficult to measure its activation. After activation factor XI becomes irreversibly complexed to a serine protease inhibitor. The most important inhibitors of factor XIa are C1inhibitor and α1-antitrypsin. Most of the factor XIa becomes complexed to C1 inhibitor but because of the longer half-life time the factor XIa-α1-antitrypsin complexes are also a good parameter to assess factor XI activation in clinical samples.64,65
Using an improved ELISA which measures factor XIa-a1-antitrypsin complexes Komiyama et al have shown activation of factor XI in 4 patients with DIC as high as 295 ng/ml (Å1,8 nM) which corresponds with activation of 5% of the total factor XI.66 This was also observed by Wuillemin et al who found elevated factor XIa-α1-antitrypsin complexes in 6 patients with DIC.65 Moreover, in a group of 13 children with meningococcal septic shock and DIC, factor XIa-α1-antitrypsin complexes were elevated in all patients and factor XIa-C1inhibitor complexes in 9 patients.67
An increase in the TAFI plasma concentration is associated with an increased formation of activated TAFI by thrombin, since the Michaelis-Menten constant for TAFI activation by thrombin is far above the TAFI plasma concentration. In mice and rats, TAFI was identified as an acute phase reactant, which suggests that increased TAFI levels during inflammation contribute to the antifibrinolytic state that is characteristic for DIC.68,69 Furthermore, plasma levels of soluble thrombomodulin fragments are elevated in patients with DIC and in vitro studies have shown that also soluble thrombomodulin fragments can stimulate TAFI activation.45 However, since information of TAFI activation in patients with DIC is lacking, the role of TAFI in the pathofysiology of DIC remains to be investigated.
Conclusion
Although clinical data are not abundant, it is reasonable to state that factor XI is activated in patients with DIC. This activation is mediated by thrombin, which is generated through the extrinsic pathway. By activation of factor XI more thrombin will be formed and this thrombin is important in the activation of TAFI (Fig. 7). The significance of TAFI in the pathogenesis DIC is not clear and awaits further research.
The role of the intrinsic pathway (factor XI, factor IX and factor VIII) as the pathway important for the maintenance of coagulation has started new research for interventions in thrombotic complications. Current antithrombotic strategies are aimed at factor Xa and/or thrombin and carry the risk of hemorrhage. Inhibiting activation of factor XI could probably result in clots that are less thrombogenic and more susceptible to fibrinolysis, without inhibiting tissue factor-dependent hemostasis. In a murine model of intracerebral stroke it was demonstrated that inhibiting factor IXa activity reduced microvascular fibrin deposition, increased cerebral perfusion and reduced the infarct volumes. This could also be achieved by giving t-PA or heparin to the mice but these interventions significantly enhanced intracerebral hemorrhage.70 Especially in patients with DIC and high risk on bleeding these kind of highly tailored interventions may turn out to be very promising.
References
- 1.
- Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. J Biol Chem. 1991;266:7353–7358. [PubMed: 2019570]
- 2.
- Gailani D, Broze GJ. Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–911. [PubMed: 1652157]
- 3.
- Saito H. Contact factors in health and disease. Semin Thromb Hemost. 1987;13:36–49. [PubMed: 3551076]
- 4.
- Kitchens CS. Factor XI: A review of its biochemistry and deficiency. Semin Thromb Haemost. 1991;17:55–72. [PubMed: 2047881]
- 5.
- Broze GJ, Girard TJ, Novotny WF. Regulation of coagulation by a multivalent Kunitz-type inhibitor. Biochemistry. 1991;29:7539–7546. [PubMed: 2271516]
- 6.
- von dem Borne P A K, Bajzar L, Meijers J C M, Nesheim ME, Bouma BN. Thrombin-mediated activation of factor XI results in a thrombin-activatable fibrinolysis inhibitor-dependent inhibition of fibrinolysis. J Clin Invest. 1997;99:2323–2327. [PMC free article: PMC508069] [PubMed: 9153272]
- 7.
- Bouma BN, Vlooswijk R A A, Griffin JH. Immunologic studies of human coagulation factor XI and its complex with high molecular weight kininogen. Blood. 1983;62:1123–1131. [PubMed: 6626744]
- 8.
- Bouma BN, Griffin JH. Human Blood Coagulation Factor XI, purification, properties, and mechanism of activation by activated factor XII. J Biol Chem. 1977;252:6432–6437. [PubMed: 893417]
- 9.
- Fujikawa K, Chung DW, Hendrickson LE, Davie EW. Amino acid sequence of human factor XI, a blood coagulation factor with four tandem repeats that are highly homologous with plasma prekallikrein. Biochemistry. 1986;25:2417–2424. [PubMed: 3636155]
- 10.
- van der Graaf F, Greengard JS, Bouma BN, Kerbiriou DM, Griffin JH. Isolation and functional characterization of the active light chain of activated human blood coagulation factor XI. J Biol Chem. 1983;258:9669–9675. [PubMed: 6604052]
- 11.
- Baglia FA, Jameson BA, Walsh PN. Fine mapping of the high molecular weight kininogen binding site on blood coagulation factor XI through the use of rationally designed synthetic analogs. J Biol Chem. 1992;267:4247–4252. [PubMed: 1740464]
- 12.
- Baglia FA, Walsh PN. A binding site for thrombin in the apple 1 domain of factor XI. J Biol Chem. 1996;271:3652–3658. [PubMed: 8631976]
- 13.
- Baglia FA, Badellino KO, Ho DH, Dasari VR, Walsh PN. A binding site for the kringle II domain of prothrombin in the apple 1 domain of factor XI. J Biol Chem. 2000;275:31954–31962. [PubMed: 10924522]
- 14.
- Baglia FA, Jameson BA, Walsh PN. Identification and chemical analysis of a substrate-binding site for factor IX in coagulation factor XIa. J Biol Chem. 1991;266:24190–24197. [PubMed: 1748688]
- 15.
- Sinha D, Seaman FS, Walsh PN. Role of calcium ions and the heavy chain of factor XIa in the activation of human coagulation factor IX. Biochemistry. 1987;26:3768–3775. [PubMed: 3498513]
- 16.
- Sun Y, Gailani D. Identification of a factor IX binding site on the third apple domain of activated factor XI. J Biol Chem. 1996;271:29023–29028. [PubMed: 8910554]
- 17.
- Baglia FA, Jameson BA, Walsh PN. Identification and characterization of a binding site for platelets in the apple 3 domain of coagulation factor XI. J Biol Chem. 1995;270:6734–6740. [PubMed: 7896818]
- 18.
- Ho DH, Badellino K, Baglia FA, Walsh PN. A binding site for heparin in the Apple 3 domain of factor XI. J Biol Chem. 1998;273:16382–16390. [PubMed: 9632702]
- 19.
- Sun M, Zhao M, Gailani D. Identification of aminoacids in the factor XI apple 3 domain required for activation of factor IX. J Biol Chem. 1999;274:36373–36378. [PubMed: 10593931]
- 20.
- Baglia FA, Jameson BA, Walsh PN. Identification and characterization of a binding site for factor XIIa in the apple 4 domain of coagulation factor XI. J Biol Chem. 1993;268:3838–3844. [PubMed: 8440679]
- 21.
- Meijers J C M, Mulvihill ER, Davie EW, Chung DW. Apple four in human blood coagulation factor XI mediates dimer formation. Biochemistry. 1992;31:4680–4684. [PubMed: 1581318]
- 22.
- McMullen BA, Fujikawa K, Davie EW. Location of the disulfide bonds in human coagulation factor XI: The presence of tandem apple domains. Biochemistry. 1991;30:2056–2060. [PubMed: 1998667]
- 23.
- Kurachi K, Davie EW. Activation of human factor XI (plasma thromboplastin antecedent) by factor XIIa (activated Hageman factor). Biochemistry. 1977;16:5831–5839. [PubMed: 588558]
- 24.
- Pixley RA, de la Cadena R, Page JD, Kaufman N, Wyshock EG, Chang A. et al. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia. J Clin Invest. 1993;91:61–68. [PMC free article: PMC329995] [PubMed: 7678610]
- 25.
- Liu CY, Nossel HL, Kaplan KL. The binding of thrombin by fibrin. J Biol Chem. 1979;254:10421–10425. [PubMed: 489603]
- 26.
- Weitz JI, Hudoba M, Massel D, Maraganore J, Hirsh J. Clot-bound thrombin is protected from inhibition by heparin-antithrombin III but is susceptible to inactivation by antithrombin III-independent inhibitors. J Clin Invest. 1990;86:385–391. [PMC free article: PMC296739] [PubMed: 2384594]
- 27.
- Gailani D, Broze G J Jr. Effects of glycosaminoglycans on factor XI activation by thrombin. Blood Coagul Fibrinol. 1993;4:15–20. [PubMed: 8457645]
- 28.
- Walsh PN, Sinha D, Koshy A, Seaman FS, Bradford H. Functional characterization of platelet-bound factor XIa: Retention of factor XIa activity on the platelet surface. Blood. 1986;68:225–230. [PubMed: 3487355]
- 29.
- Baglia FA, Walsh PN. Prothrombin is a co-factor for the binding of factor XI to the platelet surface and for platelet-mediated factor XI activation by thrombin. Biochemistry. 1998;37:2271–2281. [PubMed: 9485373]
- 30.
- Baglia FA, Walsh PN. Thrombin-mediated feedback activation of factor XI on the activated platelet surface is preferred over contact activation by factor XIIa or factor XIa. J Biol Chem. 2000;275:20514–20519. [PubMed: 10781579]
- 31.
- Scott CF, Colman RW. Fibrinogen blocks the autoactivation and thrombin-mediated activation of factor XI on dextran sulfate. Proc Natl Acad Sci USA. 1992;89:11189–11193. [PMC free article: PMC50515] [PubMed: 1454798]
- 32.
- von dem Borne P A K, Koppelman SJ, Bouma BN, Meijers J C M. Surface independent factor XI activation by thrombin in the presence of high molecular weight kininogen. Thromb Haemost. 1994;72:397–402. [PubMed: 7855791]
- 33.
- von dem Borne P A K, Meijers J C M, Bouma BN. Effect of heparin on the activation of factor XI by fibrin-bound thrombin. Thromb Haemost. 1996;76:347–353. [PubMed: 8883269]
- 34.
- Gailani D, Broze GJ. Factor XII-independent activation of factor XI in plasma: Effects of sulfatides on tissue factor-induced coagulation. Blood. 1993;82:813–819. [PubMed: 8338946]
- 35.
- von dem Borne P A K, Meijers J C M, Bouma BN. Feedback activation of factor XI by thrombin in plasma results in additional formation of thrombin that protects fibrin clots from fibrinolysis. Blood. 1995;86:3035–3042. [PubMed: 7579397]
- 36.
- Bajzar L, Nesheim M. The effect of activated protein C on fibrinolysis in cell-free plasma can be attributed specifically to attenuation of prothrombin activation. J Biol Chem. 1993;268:8608–8616. [PubMed: 8473306]
- 37.
- Bajzar L, Manuel R, Nesheim ME. Purification and characterization of TAFI, a thrombin-activatable fibrinolysis inhibitor. J Biol Chem. 1995;270:14477–14484. [PubMed: 7782309]
- 38.
- Eaton DL, Malloy BE, Tsai SP, Henzel W, Drayna D. Isolation, molecular cloning, and partial characterization of a novel carboxypeptidase B from human plasma. J Biol Chem. 1991;266:21833–21838. [PubMed: 1939207]
- 39.
- Wang W, Hendriks DF, Scharpe SS. Carboxypeptidase U, a plasma carboxypeptidase with high affinity for plasminogen. J Biol Chem. 1994;269:15937–15944. [PubMed: 8195249]
- 40.
- Bajzar L, Nesheim ME, Tracy PB. The profibrinolytic effect of activated protein C in clots formed from plasma is TAFI-dependent. Blood. 1996;88:2093–2100. [PubMed: 8822928]
- 41.
- Mosnier LO, von dem Borne P A K, Meijers J C M, Bouma BN. Plasma TAFI levels influence the clot lysis time in healthy individuals in the presence of an intact intrinsic pathway of coagulation. Thromb Haemost. 1998;80:829–835. [PubMed: 9843179]
- 42.
- Stromqvist M, Schatteman KA, Leurs J, Verkerk M, Andersson JO, Johansson T. et al. Immunological assay for the determination of procarboxypeptidase U antigen levels in human plasma. Thromb Haemost. 2001;85:12–17. [PubMed: 11204563]
- 43.
- Bajzar L, Morser J, Nesheim M. TAFI, or plasma carboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem. 1996;271:16603–16608. [PubMed: 8663147]
- 44.
- Bajzar L, Nesheim M, Morser J, Tracy PB. Both cellular and soluble forms of thrombomodulin inhibit fibrinolysis by potentiating the activation of thrombin-activatable fibrinolysis inhibitor. J Biol Chem. 1998;273:2792–2798. [PubMed: 9446587]
- 45.
- Hosaka Y, Takahashi Y, Ishii H. Thrombomodulin in human plasma contributes to inhibit fibrinolysis through acceleration of thrombin-dependent activation of plasma carboxypeptidase B. Thromb Haemost. 1998;79:371–377. [PubMed: 9493593]
- 46.
- Mosnier LO, Meijers J C M, Bouma BN. Regulation of fibrinolysis in plasma by TAFI and protein C is dependent on the concentration of thrombomodulin. Thromb Haemost. 2001;85:5–11. [PubMed: 11204587]
- 47.
- Kokame K, Zheng X, Sadler JE. Activation of thrombin-activatable fibrinolysis inhibitor requires epidermal growth factor-like domain 3 of thrombomodulin and is inhibited competitively by protein C. J Biol Chem. 1998;273:12135–12139. [PubMed: 9575159]
- 48.
- Wang W, Nagashima M, Schneider M, Morser J, Nesheim ME. Elements of the primary structure of thrombomodulin required for efficient TAFI activation. J Biol Chem. 2000;275:22942–22947. [PubMed: 10801821]
- 49.
- Sakharov DV, Plow EF, Rijken DC. On the mechanism of the antifibrinolytic activity of plasma carboxypeptidase B. J Biol Chem. 1997;272:14477–14482. [PubMed: 9162090]
- 50.
- Wang W, Boffa MB, Bajzar L, Walker JB, Nesheim ME. A study of the mechanism of inhibition of fibrinolysis by activated thrombin-activatable fibrinolysis inhibitor. J Biol Chem. 1998;273:27176–27181. [PubMed: 9765237]
- 51.
- Boffa MB, Wang W, Bajzar L, Nesheim ME. Plasma and recombinant thrombin-activatable fibrinolysis inhibitor (TAFI) and activated TAFI compared with respect to glycosyllation, thrombin/thrombomodulin-dependent activation, thermal stability, and enzymatic properties. J Biol Chem. 1998;273:2127–2135. [PubMed: 9442053]
- 52.
- Lawson JH, Kalafatis M, Stram S, Mann KG. A model for the tissue factor pathway to thrombin. I.an empirical study. J Biol Chem. 1994;269:23357–23366. [PubMed: 8083241]
- 53.
- Cawthern KM, van 't Veer C, Lock JB, DiLorenzo ME, Branda RF, Mann KG. Blood coagulation in hemophilia A and hemophilia C. Blood. 1998;91:4581–4592. [PubMed: 9616154]
- 54.
- Broze GJ, Higuchi DA. Coagulation-dependent inhibition of fibrinolysis: Role of Carboxypeptidase-U and the premature lysis of clots from hemophilic plasma. Blood. 1996;88:3815–3823. [PubMed: 8916945]
- 55.
- Bouma BN, Mosnier LO, Meijers J C M, Griffin JH. Factor XI dependent and independent activation of thrombin activatable fibrinolysis inhibitor (TAFI) in plasma associated with clot formation. Thromb Haemost. 1999;82:1703–1708. [PubMed: 10613658]
- 56.
- Minnema MC, Friederich PW, Levi M, von dem Borne P A K, Mosnier LO, Meijers J C M. et al. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI: In vivo evidence for a role of factor XI as an anti- fibrinolytic factor. J Clin Invest. 1998;101:10–14. [PMC free article: PMC508534] [PubMed: 9421460]
- 57.
- Redlitz A, Nicolini FA, Malycky JL, Topol EJ, Plow EF. Inducible carboxypeptidase activity. A role in clot lysis in vivo. Circulation. 1996;93:1328–1330. [PubMed: 8641019]
- 58.
- Klement P, Liao P, Bajzar L. A novel approach to arterial thrombolysis. Blood. 1999;94:2735–2743. [PubMed: 10515877]
- 59.
- Nerme V, Akerblom B, Legnehed A, Scharpe S, Hendriks D. Carboxypeptidase U inhibitors increase the rate of fibrinolysis in a rat DIC model Fibrinolysis & Proteolysis 200069(abstract).
- 60.
- Muto Y, Suzuki K, Ishii H. TAFIa inhibitors enhance fibrinolysis in tissue factor induced rat DIC model Fibrinol Proteol 200070 (abstract).
- 61.
- Minnema MC, Pajkrt D, Wuillemin WA, Roem D, Bleeker WK, Levi M. et al. Activation of clotting factor XI, without detectable contact activation in experimental human endotoxemia. Blood. 1998;92:3294–3301. [PubMed: 9787166]
- 62.
- Saito H, Goldsmith GH. Plasma Thromboplastin Antecedent (PTA, Factor XI): a specific and sensitive radioimmunoassay. Blood. 1977;50:377–385. [PubMed: 884316]
- 63.
- Vaziri ND, Toohey J, Powers D, Keegan K, Gupta A, Alikhani S. et al. Activation of intrinsic coagulation pathway in pre-eclampsia. Am J Med. 1986;80:103–107. [PubMed: 3079949]
- 64.
- Wuillemin WA, Minnema MC, Meijers J C M, Roem D, Eerenberg A J M, Nuijens JH. et al. Inactivation of factor XIa in human plasma assessed by measuring factor XIa-protease inhibitor complexes: Major role for C1-inhibitor. Blood. 1995;85:1517–1526. [PubMed: 7534133]
- 65.
- Wuillemin WA, Hack CE, Bleeker WK, Biemond BJ, Levi M, ten Cate H. Inactivation of factor XIa in vivo: Studies in chimpanzees and in humans. Thromb Haemost. 1996;76:549–555. [PubMed: 8902995]
- 66.
- Komiyama Y, Masuda M, Murakami T, Egawa H, Nishikado H, Murata K. New rapid assay for factor XIa-a1antitrypsin complex. Application to DIC. Thromb Res. 1989;55:527–536. [PubMed: 2814942]
- 67.
- Wuillemin WA, Fijnvandraat K, Derkx B H F, Peters M, Vreede W, ten Cate H. et al. Activation of the intrinsic pathway of coagulation in children with meningococcal septic shock. Thromb Haemost. 1995;74:1436–1441. [PubMed: 8772216]
- 68.
- Sato T, Miwa T, Akatsu H, Matsukawa N, Obata K, Okada N. et al. Pro-carboxypeptidase R is an acute phase protein in the mouse, whereas carboxypeptidase N is not. J Immunol. 2000;165:1053–1058. [PubMed: 10878383]
- 69.
- Kato T, Akatsu H, Sato T, Matsuo S, Yamamoto T, Campbell W. et al. Molecular cloning and partial characterization of rat procarboxypeptidase R and carboxypeptidase N. Microbiol Immunol. 2000;44:719–728. [PubMed: 11021404]
- 70.
- Choudhri TF, Hoh BL, Prestigiacomo CJ, Huang J, Kim LJ, Schmidt AM. et al. Targeted inhibition of intrinsic coagulation limits cerebral injury in stroke without increasing intracerebral hemorrhage. J Exp Med. 1999;190:91–99. [PMC free article: PMC2195562] [PubMed: 10429673]
Publication Details
Author Information and Affiliations
Authors
Monique C. Minnema and Joost C.M. Meijers.Copyright
Publisher
Landes Bioscience, Austin (TX)
NLM Citation
Minnema MC, Meijers JCM. Factor XI, TAFI and DIC. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.