

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Microarray analysis has yet to be widely accepted for diagnosis and classification of human cancers, despite the exponential increase in microarray studies reported in the literature. Among several methods available, a few refined approaches have evolved for the analysis of microarray data for cancer diagnosis. These include class comparison, class prediction and class discovery. Using as examples some of the major experimental contributions recently provided in the field of both hematological and solid tumors, we discuss the steps required to utilize microarray data to obtain general and reliable gene profiles that could be universally used in clinical laboratories. As we show, microarray technology is not only a new tool for the clinical lab but it can also improve the accuracy of the classical diagnostic techniques by suggesting novel tumor-specific markers. We then highlight the importance of publicly available microarray data and the development of their integrated analysis that may fulfill the promise that this new technology holds for cancer diagnosis and classification.
Introduction
Current cancer classification includes more than 200 types of cancer. For the patient to receive appropriate therapy, the clinician must identify as accurately as possible the cancer type. Although analysis of morphologic characteristics of biopsy specimens is still the standard diagnostic method, it gives very limited information and clearly misses much important tumor aspects such as rate of proliferation, capacity for invasion and metastases, and development of resistance mechanisms to certain treatment agents. To appropriately classify tumor subtypes, therefore, molecular diagnostic methods are needed. The classical molecular methods look for the DNA, RNA or protein of a defined marker that is correlated with a specific type of tumor and may or may not give biological information about cancer generation or progression. However, a major advantage of microarray is the huge amount of molecular information that can be extracted and integrated to find common patterns within a group of samples. As we will show here, microarrays could be used in combination with other diagnostic methods to add more information about the tumor specimen by looking at thousands of genes concurrently. This new method is revolutionizing cancer diagnostics because it not only classifies tumor samples into known and new taxonomic categories, and discovers new diagnostic and therapeutic markers, but it also identifies new subtypes that correlate with treatment outcome.
Revealing Expression Profiles for Cancer Diagnosis and Classification
Data Analysis: Supervised and Unsupervised Methods
Each microarray experiment generates thousands of data points and reports are written in a dense technical jargon. It is easy to feel lost when trying to make sense of it all. For this reason, it is important to clearly define certain technical terms as well as goals of microarray experiments. To understand how microarrays are used, the jargon “class” and, more specifically, “known class” must be first defined. A class refers to any characteristic shared by one group of samples but not other samples: e.g., cancer versus normal tissue, metastatic versus primary tumor, responders to cancer treatment versus nonresponders. A “known class” is any differentiating characteristic that the researcher will use to label the tumor samples under study a priori the data analysis. The two main goals of microarray studies are: 1) to identify molecular signatures associated with known classes, and 2) to discover new classes. To achieve those goals, two different approaches to data analysis are taken, the Supervised method (first goal above) and the Unsupervised method (second goal)1 (Fig. 1). To read and understand microarray-based studies, knowledge of these different methods, will greatly help to understand the authors' hypothesis and data interpretation.
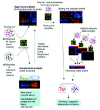
Supervised methods of analysis are used predominantly to identify the differences at molecular level between known classes (Class Comparison) and to diagnose or “predict” to which class a new tumor sample belongs (Class Prediction). By contrast, in unsupervised methods, the samples are not labeled to belong to different clinicopathologic classes before data analysis (i.e., “unknown class”). When the purpose of the experiment is to test the hypothesis that the samples are composed of different classes, the approach is called Class Discovery. Class Discovery attempts to identify new sub-classes of tumors in cancers where the actual classification needs more definition: for instance, when the classification does not explain the different patient survival after cancer treatment.
In Class Comparison studies, the purpose is to understand the differences in gene expression that might be responsible for the differences between compared classes of tumors and to, perhaps, find hints on the genes that might deserve further study. In cancer diagnosis, however, Class Comparison is usually incorporated into Class Prediction.
Class Prediction studies build a gene “predictor” based on the genes whose expression differs between the different classes of tumors under study. A predictor is a gene expression-based multivariate function that will use the genes identified in Class Comparison to assign new tumor sample(s) into the correct class (Fig. 1). However, this method suffers from one major limitation called over fitting (1). This means that the classification algorithm performs well on the samples from which it was built but poorly on independent samples. Therefore, the validation of the gene predictor is necessary for future clinical applications. The ideal predictor is built with a “training” group of samples and then validated on a “test” group of samples.2 Moreover, samples in the test and training groups should be preferentially collected and analyzed at different time points in order to ensure independency between them and to validate the predictor in similar conditions as it might be applied in the future. An important caution should be taken into account. Since the samples are a priori classified based on the currently accepted diagnostic tests, which are neither 100% sensitive nor specific, this may decrease the accuracy of the gene predictor by including in the training set a few misclassified samples. Some good examples of this type of study are discussed below.3-5
In Class Discovery studies, the samples are grouped depending on their global gene expression without reference to tumor type, grade or any other characteristic. It analyzes the expression of thousands of genes to try to discover new taxonomic groups within the samples (Fig. 1). As an unsupervised method, it will uncover the predominant relationship of the samples' gene signature, which not always corresponds with the potential clinically relevant relationship. Examples of this kind of studies are discussed later in the chapter.3,5-9
Examples from the Literature
Over the last few years, many studies have focused on the classification and diagnosis of cancer using microarray technology. Here, we will discuss a few examples that show how this technique can improve on the information given by classical diagnostic methods. The first three studies are in hematological malignancies, for a more extended review on the microarray advances in this area you can read Eber and Golub,10 and the next four studies are in solid tumors.
Hematological Malignancies
The initial microarray studies were focused on hematological tumors for two main reasons: 1) purification of certain cell populations from the tumor samples is easy according to cell surface markers (for instance, Alizadeh et al purified chronic lymphocyte leukaemia cells by using CD19, a B cell marker) or through Ficoll sedimentation to obtain mononuclear cells from peripheral blood or bone marrow specimens3,5 and 2) wide knowledge of hematopoiesis and its genetic regulation helped to understand the complicated gene expression data generated by microarrays.
Example of Class Prediction
In 1999, a pioneer study analyzed 38 bone marrow samples from acute leukemia patients.3 Acute leukemia can be divided into two groups: acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). The problem is that for such diagnosis, several diagnostic techniques need to be run because no single one is currently sufficient and even then, the diagnosis is not always correct. The authors used supervised analysis for Class prediction to come up with a list of 50 genes that were differentially expressed between the initial 27 ALL and 11 AML training samples. Then, they apply the predictor of 50 genes to a test set of 34 new leukemia samples independently collected from the training samples. Twenty-nine of the 34 samples that formed the test set were correctly classified, supporting the possibility that in the future gene predictors obtained from larger training set of samples, could be used to supplement existing diagnostic methods. Many of the genes that formed the predictor set encoded for proteins important for cell cycle, cell adhesion, transcription or oncogenes, which could give insights into cancer pathogenesis and pharmacology as well as having diagnostic value. As a second goal of this study, the authors used Class discovery method on the initial 38 leukemia samples to see whether global gene expression analysis could have distinguished between AML and ALL if these two diagnostic classes would not have been known a priori. By using self-organizing maps (SOM), where the user specifies the number of classes to be identified, and setting them in two, 24 of the 25 ALL samples were cluster together in one group and 10 of the 13 AML samples were clustered in the second one. This showed that Class discovery studies are able to uncover diagnostic classes of tumor in cases when morphological or phenotypical tests are not, although biological and clinical information seemed necessary to interpret the results.
Examples of Class Discovery: The Basis for Predicting Prognosis
The next two studies5,7 are good examples of how Class discovery approach is able to resolve new taxonomic subclasses. The discovery of new classes, when added to clinical information linked to them (as it is survival after treatment), can give very important additional prognostic information.
Alizadeh et al7 studied large B-cell lymphoma (DLBCL), the most common subtype of nonHodgkin's lymphoma, for which there are not reliable morphological, clinical, immunohistochemical or genetic diagnostic markers to recognize possible subclasses.11 By using unsupervised methods for Class discovery on samples from 40 DLBCL patients, the authors were able to distinguish two previously unknown groups of DLBCL. The two groups were called “germinal center B-like DLBCL” and “activated B-like DLBCL” because the main differences between them were genes involved in B cell activation and in germinal center formation. These two new taxonomic groups are not only biologically relevant, but they also have an important prognostic value, as the authors showed that five years after anthracycline-based chemotherapy treatment, 76% of germinal center B-like DLBCL patients survived, while only 16% of activated B-like DLBCL did.
More recently, Bullinger et al5 made a larger scale study on 116 samples from adults with AML including 45 with normal karyotype. Even though karyotype abnormalities are the most powerful prognostic factor in AML patients,12,13 35% to 50% of patients showing a normal karyotype have an unpredictable prognosis. Class discovery analysis of all the AML samples divided them into new molecular subclasses. Interestingly, the 45 patients with normal karyotype were divided in two groups that were found to have different survival rates. The authors then built a 133 genes predictor that was able to differentiate among patients with normal karyotype into good and poor prognosis. This study was the first one able to do so in AML patients with normal karyotype. Although the initial purpose of this study was the Class discovery of new subtypes of AML, the complementary clinical information on survival rates allowed the additional prognostic value to the new AML classification.
Solid Tumors
Solid tumor biopsies not only contain malignant cells, but may also contain different percentages of fibroblasts, endothelial, and immune cells that will influence the mRNA pool of the sample. Therefore, it was thought that the heterogeneity of cell types within the biopsies would not allow for “clean” cancer specific genetic studies.14 For this reason, some authors preferred to purify the tumor cells from the biopsy by laser capture microdisection15 before doing gene expression studies. Some studies have shown, however, that the data obtained from the whole tumor is very similar to the data obtained by laser microdisected tumor cells from the same specimens.16,17 Besides, nonmalignant cells in the tumor microenvironment may play a role in tumor formation, response to treatment and metastases formation.18-20 By purifying only malignant cells from the tumor biopsies, not only this information will be lost but it will also increase the cost and time of the procedure, making harder for microarrays to be implemented as a regularly used clinical diagnostic method. The studies below prove that purification of malignant cells might not be necessary to obtain reliable and useful diagnostic information from solid tumors.
Examples of Class Prediction: Improving Treatment Decisions
Gene expression analysis proved able to detect the metastatic potential of primary tumors.4 In this work, 12 metastatic adenocarcinoma nodules of diverse origin (lung, breast, prostate, colorectal, uterus, and ovary) were compared with 64 primary adenocarcinomas representing the same tumor types from different individuals, forming a training set of 76 samples. The authors found 128 genes differentially expressed between the metastatic and the primary tumors and use these genes to build a predictor that was tested to classify primary tumors of different origins (62 lung adenocarcinomas, 78 primary breast adenocarcinomas, 21 prostate adenocarcinomas, 60 medulloblastomas). They found that all the previous tumors were divided into two classes depending on how similar their molecular profile were to the metastases one. The conclusion of the study is that primary tumors carrying the metastases-like gene expression signature were associated with metastasis and worse clinical outcome. Another interesting feature of this work is that the authors used data developed from different laboratories on different array platforms to test their 128 genes predictor.
In a more recent work that analyzed primary head and neck squamous cell carcinomas (HNSCCs) Roepman et al9 were able to build a gene predictor that could detect local lymph node metastases using material from primary HNSCCs. The predictor, formed by 102 genes, outperformed current clinical diagnostic methods with an overall predictive accuracy of 86%, while the current diagnostic method had 68%. This improvement in the diagnosis has a lot of relevance for treatment selection and the authors estimated that by using microarrays to diagnose the existence of local metastases, 75% of patients that were really metastasis-free but diagnosed as carrying possible metastases, could have avoided radical neck dissection treatment. This work also presents interesting biological information about the genes differentially expressed between the two classes of primary tumors compared here: those with local metastases and those without local metastases. Interestingly, half of the 102 genes that formed the predictor have unknown role in metastases formation and could give more insights into how this process occurs.
Examples of Class Discovery
As an example of class discovery study, Bittner et al6 were able to identify previously unrecognized subtypes of cutaneous melanoma by gene expression studies of 31 melanoma biopsies. The authors found a group of 19 melanoma tumors that clustered together showing strong similarities at molecular level. Despite the lack of statistical association of this group of melanoma samples with any clinical variable, they showed that samples within this group had reduced motility and invasiveness in in vitro tests respect to samples that didn't belong to the group. This was a nice attempt to use gene expression profiling for the generation of melanoma taxonomy, however it shows the difficulties of doing so when such taxonomy is not linked to easily detectable clinical differences.
Another Class discovery study8 was able to differentiate 4 sub-groups of breast tumors: estrogen receptor positive/luminal-like, basal-like, Erb-B2 positive and normal breast tissue-like, when separating a total of 65 samples according to the expression of 496 genes. Interestingly, the four subtypes were not visible in a first analysis of their data, when they looked at a larger number of genes. The reason for this was the use of different gene selection criteria. The first list of 1,753 genes was based on the assumption that all the samples were independent between each other. However, there were 20 pair-wise comparisons of the same tumors before and after chemotherapy. When trying to group the 65 samples according to their global gene expression, the similarities between the samples coming from the same tumor overcame the similarities between the samples coming from a hypothetical same tumor subtype. Results from this first analysis showed the need to treat pair wise samples as if they belonged to the same tumor subtype and look for other samples that had similar gene expression. These biological criteria were used to create a second list of 496 genes that revealed the 4 breast cancer subtypes; this is an example of how biologist and statistician must work together to resolve the intricacy of gene expression analysis. This new classification of breast tumors has been supported by a follow up study.21
Overall, microarrays have a remarkable potential as a new diagnostic tool in oncology showing substantial improvements over conventional diagnostic and classification criteria for many different types of tumors. Better diagnosis will improve the decision making process to choose the right treatment. Better classification, when combined with treatment response data, will improve cancer prognosis.
Using Expression Profiles in the Clinic
How to Apply a Published Microarray Class Predictor to Classify New Samples
Despite great advances in discovering cancer molecular profiles, the proper application of microarray technology to routine clinical diagnostics is still unresolved. One key limitation is that an individual tumor cannot be classified independently. It needs to be compared to other samples or “standards”, whose classification is known, and which are analyzed under the same conditions as the individual tumor. For this, some points appear to be critical. First, if the predictor was created in the same lab as the sample of interest being analyzed, the sample preparation, array set up, reference sample (for two-color design), slide processing and analysis should be exactly the same as for the original set. The major limitation here might be the availability of the same reference sample. When using one-color design, it is not necessary to use reference sample for hybridization, but all other cautions are essential for the correct classification. Second, if one wants to apply predictor genes discovered in another lab, then the task is more complex. In order to obtain comparable results, usage of the originally established protocol is essential. Recently the question of interlaboratory comparability was addressed for microarray data on human tumor specimens.22 This work showed that, under similar technical conditions, a high correlation between gene expressions in repeated samples could be obtained regardless of the laboratory in which the experiments were done. However, even when using the same protocol and microarray platform, it is still necessary to analyze a set of known samples together with the unknown one/s. Furthermore, a large number of samples from several independent datasets are required to guarantee the applicability of the validated profiles.23 Although demanding, the application of a molecular profile (previously described in one laboratory) by a second laboratory with a slightly different framework may represent an important benefit. In fact, it helps to define how general or specific to certain situation/s the profile is. For example, a set of 231 genes was described by van't Veer et al24 as discriminating for prognosis in node-negative untreated breast cancer patients. However, a different laboratory found that a subset of 93 genes, out of the 231 genes forming the predictor, was valid to make the same discrimination even in a more heterogeneous population of node-positive/negative patients treated with adjuvant therapy.25
Translation of microarray profiles into clinical practice is already beginning in some academic centers in the Netherlands and United States and profiles that have been validated in retrospective studies are now being applied in prospective clinical trials.26
How to Select Biomarkers from a Microarray Class Predictor
Another way to explore genome-wide expression data for cancer diagnosis is to translate this information into surrogate molecular markers (Fig. 2). There are at least two important advantages for doing this. First, they can be measured by relatively cheap and widely used clinical methods such as RT-PCR, ELISA and immunohistochemistry. Second, they can be detected in serum or other body fluids permitting the establishment of noninvasive diagnostic test, which is very important especially in the cases of cancers with more difficult access for diagnostic biopsy (e.g., lung, ovary, pancreas). Usually, the biomarkers will be chosen from the list of genes that form a predictor. But genes that, when combined, were good predicting the class to which a new sample belongs are not necessary good biomarkers when used alone or combined with just a few other genes. Microarray predictors usually consist of tens or hundreds of molecules. Therefore, two of the main questions for translation of microarray classifiers into diagnostic markers are: first, which genes should be selected from microarray profiles and second, how to select the minimum number of these genes sufficient for good diagnostic classification. Although several statistical procedures were suggested for this purpose,27,28 currently there is no consensus about the best one. Apparently, the use of multiple algorithms increases the confidence and validity of the selected genes.28
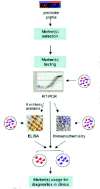
At the moment, real time RT-PCR is the most widely used technique for validation of microarray results as well as for attempting to substitute the microarray profiles for diagnostic markers. It is important to remember that, despite its similarity to microarray measurement, RT-PCR could give slightly different results. A factor that greatly contributes to the difference is the normalization procedure, which is much more precise in microarrays than in RT-PCR. In fact, microarrays generally use global normalization including all genes expressed by the sample (usually a few thousands) since the majority of the genes don't show significant expression variation across all the samples. Consequently, the normalization is not influenced as much (as it is in RT-PCR) when one or a few control (or housekeeping) genes don't behave as such and show variation on their expression among the samples.
Gordon and colleagues29 proposed an interesting solution in the form of a ratio-based method of samples classification that circumvents this problem. First, ratios between genes showing opposite expressions in the clinical groups of samples are calculated. Then, samples are assigned to one of the groups accordingly to the value of the ratio. The authors used this approach in two studies. In the first one,29 malignant pleural mesothelioma and adenocarcinoma of the lung were differentially diagnosed by means of eight genes. Five were up-regulated and three were down-regulated in mesothelioma respect to adenocarcinoma giving, therefore, 15 ratios. Any individual ratio had at least 90% of accuracy discriminating the tumor samples and they reached 99% when 3 random ratios were combined. In the second one,30 the same authors selected some genes from the published microarray data in prostate cancers,31,32 created the optimal ratio-based test and examined it using RT-PCR in an additional cohort of cancer and normal tissue. A 3-ratio test using 4 genes was 90% accurate distinguishing normal prostate and cancer samples. Thus, the most important feature of this solution is that using gene expression ratios it is possible to avoid the selection of “right” housekeeping genes and the normalization process for assignment of samples to classes.
However, even using conventional normalization of RT-PCR results, the microarray results can be translated into RT-PCR diagnostic profiles. In fact, Lossos and colleagues33 studied by RT-PCR 36 genes whose expression had been reported to predict survival in diffuse large-B-cell lymphoma based on microarray data. Six genes that were among the strongest predictors entered the multivariate model and were able to distinguish different survival groups.
Similarly, a diagnostic discrimination between benign and malignant esophageal tissue was proposed using the expression of the most informative genes selected from microarrays and further evaluated by RT-PCR.34 In this study, logistic regression and linear discriminant analysis were applied for the selection of clinically useful gene-classifiers. Continuing in the same field of gastrointestinal oncology and following up microarray experiments, Mori et al35 found highly specific markers that detected minuscule amounts of cancer cells in cytology-negative peritoneal washings by RT-PCR. Importantly, they prospectively identified a proportion of patients with minimal residual disease that could not be diagnosed and treated otherwise.
Because RT-PCR can easily and fast detect mRNA levels of considerably many genes, and it does not depend on availability of specific reagents (like antibodies), it is the most rapid translation method of microarray observations into clinical practice. ELISA and immunohistochemistry, however, detect proteins in quantitative or semi-quantitative manner. Therefore, these two methods require a more complex procedure for marker selection and validation, since not only the mRNA expression needs to be validated but also the level of the corresponding protein in tissue and or serum.
To increase a chance that marker candidates selected from large scale gene expression data will pass all rigorous validation requirements, results of microarrays from different studies could be screened. An example of a tissue marker discovered using data from several microarray datasets is alpha-methylacyl CoA racemase (AMACR).36 AMACR was selected using 4 independent datasets where the gene was over-expressed in prostate cancer comparing to benign prostate tissue. During validation in an independent set of samples, the same results were obtained when measuring mRNA and protein expression using RT-PCR and immunoblot, respectively. Then immunohistochemistry on tissue array was applied to analyze a large number of samples and evaluate clinical utility of AMACR. Interestingly, AMACR immunostaining showed not only good sensitivity (97%) and specificity (100%) for prostate cancer in the whole sample population, but it performed well also in diagnostically challenging cases that needed additional expert pathological review.
Similarly, the proteins villin and moesin were found to be tissue biomarkers that successfully distinguished between colon and ovarian adenocarcinomas.37 To do so, the authors measured gene expression and protein levels in tumor cell lines. As a result of these experiments and also based on antibody availability, villin and moesin were selected as candidates for colorectal adenocarcinoma and ovarian adenocarcinoma, respectively. Then, after sequence verification, and corroboration of mRNA expression using Affimetrix array, they validated the protein levels. This was done by protein lysate microarrays followed by immunohistochemistry on tissue microarray where the authors obtained high sensitivity and specificity for both markers. This and the AMACR studies represent good examples on how a multi-step approach including genomic, proteomic, and tissue array profiling, results in selection of very few but efficient diagnostic tissue markers.
From a diagnostic point of view, serum cancer markers are even more important than tissue markers because of their ease of procurement for large screenings for early cancer diagnosis. However, the search for serum markers is the more challenging. In fact, a candidate for serum marker, selected from gene expression profiles, should not only be over-expressed locally in the cancer microenvironment, but also codes for a protein that is secreted to the periphery in sufficient levels to be detected in blood. In this situation, bioinformatics tools like Gene Ontologies are helpful to choose genes with the characteristics of interest (e.g., secreted molecule) among the huge amount of differentially expressed genes.
Ovarian cancer is a good example of discovery of serum markers. This type of cancer is usually diagnosed in advanced stage, when only about 28% of the patients survive 5 years.38 In a series of studies, three serum diagnostic candidates (prostatin, osteopontin and creatine kinase B) were evaluated.37-39 By microarrays, the authors found these three proteins (among many others) over-expressed in ovarian tumor versus normal cell lines and the three markers were selected based on antibodies availability. After corroboration of the results by real time RT-PCR and immunohistochemistry, the authors screened sera from patients with ovarian cancer, benign disease, and healthy controls by ELISA. The results showed a strong association between increased levels of the markers and ovarian cancer. Perhaps future works combining all three markers and in a larger sample setting will show how useful these three markers could be in diagnostic screening of ovarian cancer.
Overall, microarray expression profiles are an excellent source of useful markers, which will allow the diagnosis of different tumors by conventional techniques in clinical laboratories.
Perspective
As the mass of transcriptome data for cancer diagnosis/classification continues to grow and each single study may have a limited power and validity, there is the need for combined analysis of publicly available data. To reach this goal, every new publication in this field is required to follow the MIAME (Minimum Information About a Microarray Experiment) guidelines (described in: http://www.mged.org/Workgroups/MIAME/miame_checklist.html) and to deposit its microarray data to an open database. For this purpose, two open databases are most commonly used: Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and Array Express, (http://www.ebi.ac.uk/arrayexpress/). Using this tool, some groups of researchers tried to join data from different microarray works, either to reveal new expression profiles or to select markers for diagnostic assessment by other than microarray techniques.40-42 Recently, Rhodes and colleagues41 addressed the question of microarray meta-analysis (i.e., combined analysis of the results of different microarray studies) (Fig. 1) in order to identify common gene expression signatures of human cancers. Contrary to the other studies focused on a single tissue type and model, they collected and analyzed data from more that 3,700 cancer samples representing more than 10 tissue types. A common transcriptional profile (“meta-profile”) universally activated across most cancer types compared to normal tissue was detected. In addition, more aggressive, undifferentiated cancers showed a distinct meta-signature. This work identified common features of neoplastic transformation and progression and it is a tool for searching potential universal diagnostic markers.
To reach its full potential in cancer diagnosis and classification microarray technology needs improvement of its ancillary technologies such as development of new microarray platforms, statistics and software for analysis and data mining. This will not only simplify technical and analytical procedures but will also make them more precise and cheaper. In addition, inter-laboratory cooperation for ongoing meta-profiles will help produce standardized diagnostic methods utilizing microarrays.
In conclusion, microarrays are beginning to take an important place in clinical oncology practice. Although the main potential success of microarrays is related to evaluation of patients' prognosis, microarrays also improve current clinical diagnostics, discover new diagnostic markers and identify new taxonomic classes of tumors.
Aknowledgements
We want to thank Brandon Reines for critically reading this chapter.
References
- 1.
- Simon R, Radmacher MD, Dobbin K. et al. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J Natl Cancer Inst. 2003;95:14–18. [PubMed: 12509396]
- 2.
- Ntzani EE, Ioannidis JP. Predictive ability of DNA microarrays for cancer outcomes and correlates: An empirical assessment. Lancet. 2003;362:1439–1444. [PubMed: 14602436]
- 3.
- Golub TR, Slonim DK, Tamayo P. et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. [PubMed: 10521349]
- 4.
- Ramaswamy S, Ross KN, Lander ES. et al. A molecular signature of metastasis in primary solid tumors. Nat Genetics. 2003;33:49–54. [PubMed: 12469122]
- 5.
- Bullinger L, Dohner K, Bair E. et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350:1605–1616. [PubMed: 15084693]
- 6.
- Bittner M, Meltzer P, Chen Y. et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. [PubMed: 10952317]
- 7.
- Alizadeh AA, Eisen MB, Davis RE. et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. [PubMed: 10676951]
- 8.
- Perou CM, Sorlie T, Eisen MB. et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. [PubMed: 10963602]
- 9.
- Roepman P, Wessels LF, Kettelarij N. et al. An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet. 2005;37:182–186. [PubMed: 15640797]
- 10.
- Ebert BL, Golub TR. Genomic approaches to hematologic malignancies. Blood. 2004;104:923–932. [PubMed: 15155462]
- 11.
- Harris NL, Jaffe ES, Diebold J. et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: Report of the Clinical Advisory Committee meeting. J Clin Oncol. 1999;12:3835–3849. [PubMed: 10577857]
- 12.
- Grimwade D, Walker H, Oliver F. et al. The importance of diagnostic cytogenetics on outcome in AML: Analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998;92:2322–2333. [PubMed: 9746770]
- 13.
- Bloomfield CD, Lawrence D, Byrd JC. et al. Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res. 1998;58:4173–4179. [PubMed: 9751631]
- 14.
- Player A, Barrett JC, Kawasaki ES. Laser capture microdissection, microarrays and the precise definition of a cancer cell. Expert Rev Mol Diagn. 2004;4:831–840. [PubMed: 15525225]
- 15.
- Ohyama H, Zhang X, Kohno Y. et al. Laser capture microdissection-generated target sample for high-density oligonucleotide array hybridization. Biotechniques. 2000;29:530–536. [PubMed: 10997267]
- 16.
- Ernst T, Hergenhahn M, Kenzelmann M. et al. Decrease and gain of gene expression are equally discriminatory markers for prostate carcinoma: A gene expression analysis on total and microdissected prostate tissue. Am J Pathol. 2002;160:2169–2180. [PMC free article: PMC1850818] [PubMed: 12057920]
- 17.
- Sanchez-Carbayo M, Saint F, Lozano JJ. et al. Comparison of gene expression profiles in laser-microdissected, nonembedded, and OCT-embedded tumor samples by oligonucleotide microarray analysis. Clin Chem. 2003;49:2096–2100. [PubMed: 14633888]
- 18.
- Dorudi S, Hart IR. Mechanisms underlying invasion and metastasis. Curr Opin Oncol. 1993;5:130–135. [PubMed: 7678990]
- 19.
- Basset P, Okada A, Chenard MP. et al. Matrix metalloproteinases as stromal effectors of human carcinoma progression: Therapeutic implications. Matrix Biol. 1997;15:535–541. [PubMed: 9138286]
- 20.
- Chung LW, Baseman A, Assikis V. et al. Molecular insights into prostate cancer progression: The missing link of tumor microenvironment. J Urol. 2005;173:10–20. [PubMed: 15592017]
- 21.
- Sorlie T, Tibshirani R, Parker J. et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–8423. [PMC free article: PMC166244] [PubMed: 12829800]
- 22.
- Dobbin KK, Beer DG, Meyerson M. et al. Interlaboratory comparability study of cancer gene expression analysis using oligonucleotide microarrays. Clin Cancer Res. 2005;11:565–572. [PubMed: 15701842]
- 23.
- Michiels S, Koscielny S, Hill C. Prediction of cancer outcome with microarrays: A multiple random validation strategy. Lancet. 2005;365:488–492. [PubMed: 15705458]
- 24.
- van Veer t LJ, Dai H, van de Vijver MJ. et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. [PubMed: 11823860]
- 25.
- Sotiriou C, Neo SY, McShane LM. et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci USA. 2003;100:10393–10398. [PMC free article: PMC193572] [PubMed: 12917485]
- 26.
- Kallioniemi O. Medicine: Profile of a tumour. Nature. 2004;428:379–382. [PubMed: 15042074]
- 27.
- Xiong M, Li W, Zhao J. et al. Feature (gene) selection in gene expression-based tumor classification. Mol Genet Metab. 2001;73:239–247. [PubMed: 11461191]
- 28.
- Fu LM, Fu-Liu CS. Multi-class cancer subtype classification based on gene expression signatures with reliability analysis. FEBS Lett. 2004;561:186–190. [PubMed: 15013775]
- 29.
- Gordon GJ, Jensen RV, Hsiao LL. et al. Translation of microarray data into clinically relevant cancer diagnostic tests using gene expression ratios in lung cancer and mesothelioma. Cancer Res. 2002;62:4963–4967. [PubMed: 12208747]
- 30.
- Bueno R, Loughlin KR, Powell MH. et al. A diagnostic test for prostate cancer from gene expression profiling data. J Urol. 2004;171:903–906. [PubMed: 14713850]
- 31.
- Dhanasekaran SM, Barrette TR, Ghosh D. et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–826. [PubMed: 11518967]
- 32.
- Welsh JB, Sapinoso LM, Su AI. et al. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–5978. [PubMed: 11507037]
- 33.
- Lossos IS, Czerwinski DK, Alizadeh AA. et al. Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. N Engl J Med. 2004;350:1828–1837. [PubMed: 15115829]
- 34.
- Brabender J, Marjoram P, Salonga D. et al. A multigene expression panel for the molecular diagnosis of Barrett's esophagus and Barrett's adenocarcinoma of the esophagus. Oncogene. 2004;23:4780–4788. [PubMed: 15107828]
- 35.
- Mori K, Aoyagi K, Ueda T. et al. Highly specific marker genes for detecting minimal gastric cancer cells in cytology negative peritoneal washings. Biochem Biophys Res Commun. 2004;313:931–937. [PubMed: 14706632]
- 36.
- Rubin MA, Zhou M, Dhanasekaran SM. et al. alpha-Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA. 2002;287:1662–70. [PubMed: 11926890]
- 37.
- Nishizuka S, Chen ST, Gwadry FG. et al. Diagnostic markers that distinguish colon and ovarian adenocarcinomas: Identification by genomic, proteomic, and tissue array profiling. Cancer Res. 2003;63:5243–50. [PubMed: 14500354]
- 38.
- Huddleston HG, Wong KK, Welch WR. et al. Clinical applications of microarray technology: Creatine kinase B is an up-regulated gene in epithelial ovarian cancer and shows promise as a serum marker. Gynecol Oncol. 2005;96:77–83. [PubMed: 15589584]
- 39.
- Mok SC, Chao J, Skates S. et al. Prostasin, a potential serum marker for ovarian cancer: Identification through microarray technology. J Natl Cancer Inst. 2001;93:1458–1464. [PubMed: 11584061]
- 40.
- Kim JH, Skates SJ, Uede T. et al. Osteopontin as a potential diagnostic biomarker for ovarian cancer. JAMA. 2002;287:1671–9. [PubMed: 11926891]
- 41.
- Rhodes DR, Yu J, Shanker K. et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci USA. 2004;101:9309–9314. [PMC free article: PMC438973] [PubMed: 15184677]
- 42.
- Choi JK, Choi JY, Kim DG. et al. Integrative analysis of multiple gene expression profiles applied to liver cancer study. FEBS Lett. 2004;565:93–100. [PubMed: 15135059]
- Microarrays for Cancer Diagnosis and Classification - Madame Curie Bioscience Da...Microarrays for Cancer Diagnosis and Classification - Madame Curie Bioscience Database
- Regulation of the Rod Photoreceptor Cyclic Nucleotide-Gated Channel - Madame Cur...Regulation of the Rod Photoreceptor Cyclic Nucleotide-Gated Channel - Madame Curie Bioscience Database
- Regulation of Gene Expression by Coupling of Alternative Splicing and NMD - Mada...Regulation of Gene Expression by Coupling of Alternative Splicing and NMD - Madame Curie Bioscience Database
- miRNAs need a Trim Regulation of miRNA Activity by Trim-NHL Proteins - Madame Cu...miRNAs need a Trim Regulation of miRNA Activity by Trim-NHL Proteins - Madame Curie Bioscience Database
- GSK3-Signal Regulation of Pattern Formation in Dictyostelium: Wnt-Like Pathways ...GSK3-Signal Regulation of Pattern Formation in Dictyostelium: Wnt-Like Pathways During Non-Canonical Multicellular Development - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...